Recognize the Signs of Ocular Melanoma
Key features allow you to identify melanomas early and help—even save—your patients.
Goal Statement:
The appearance of ocular melanomas make them hard to identify without the proper clinical insight, and some occur frequently enough to warrant education regarding their identification and treatment. This course provides a detailed overview of choroidal, ciliary body, iris and conjunctival melanomas, including clinical presentation, risk factors, differentials, prognostics and treatment options.
Faculty/Editorial Board:
Sara Weidmayer, OD
Credit Statement:
This course is COPE approved for 2 hours of CE credit. COPE ID: 47396-PS. Please check your state licensing board to see if this approval counts toward your CE requirement for relicensure.
Joint-Sponsorship Statement:
This continuing education course is joint-sponsored by the Pennsylvania College of Optometry.
Disclosure Statement:
Dr. Weidmayer has no financial relationships to disclose.
In the past, enucleation was the standard treatment for uveal melanoma (UM). However, it became apparent that early detection and prompt referral for treatment could reduce mortality and allow less invasive, globe-sparing treatment modalities. The role of the primary eye care provider is integral for early detection.
Uveal Melanomas
Melanomas are malignant tumors that occur when pigment-producing melanocytes abnormally over-proliferate and gain resistance to apoptosis.1,2 Ninety-five percent of ocular melanomas (OM) are uveal, while conjunctival OMs constitute the remaining 5%.2 Rarely, OM formation occurs in the eyelid or orbit.3 The uveal tract is composed of the anteriorly-located iris and the posteriorly-located ciliary body and choroid. Approximately 90% of UMs occur in the choroid.1,4,5 The remaining 9% of cases occur in the ciliary body and iris (7% and 2%, respectively).1,4,5 These can develop from a pre-existing lesion, such as a choroidal or iris nevus.1
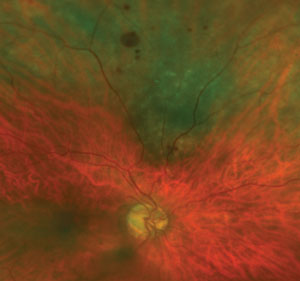 |
| This photo shows an 8mm x 6mm mottled melanotic choroidal nevus with no appreciable thickening in the superior midperiphery, with overlying drusen and intraretinal hemorrhaging. There are also intraretinal hemorrhages extending into the superior periphery. The lesion has no evidence of orange pigment or subretinal fluid. Surprisingly, this choroidal nevus has remained stable in size and thickness for several years, and is being monitored by an ocular oncologist due to its size. |
Uveal melanomas are the most common primary intraocular malignancy in adults. In fact, they are second only to childhood retinoblastoma as the most common primary intraocular malignancy.1,4,6 However, with only between four and 20 per one million cases per year globally, UM is still very rare.1,6 It has been estimated that just shy of 7,100 patients develop UM globally per year, with around 2,500 of those being North Americans. The rare conjunctival OM presents in 200 North Americans annually.3
The incidence of UM is found almost entirely in the Caucasian population—98% of cases.6,7 They can occur at any age, though the median onset is between 55 and 60 years old and only 1% of cases occur in people under 20. Iris melanomas tend to appear at a younger age. On average, an iris melanoma diagnosis occurs in the patient's early 40s.7,8 UM is slightly more prevalent in men than women.4,6,9,10
Clinical Presentation
The morphology of posterior UM varies substantially with respect to shape, size and color. Coloration can range from amelanotic (about 15% of cases) to darkly pigmented (about 55%), and the pigmentation is often mottled (about 30%).1,3,4 They may be shallow and diffuse (5%), but are often irregular solid masses that are either abruptly elevated in the form of a dome (75%), or are mushroom-shaped (20%) if they erupt through Bruch's membrane. They also may become multi-lobular with growth.1,5,11 Choroidal melanomas are grouped as small (0 to 3mm), medium (3.1 to 8mm) or large (greater than 8.1mm), based on thicknesses.3,5
Iris melanomas are pigmented more than 80% of the time, and almost half (45%) appear in the inferior quadrant.3 They may appear thin and diffuse, in a transparent "tapioca" pattern infiltrating the iris, or nodular with the appearance of feeder vessels.3
Patients with melanomas may present with a variety of symptoms, depending on the size, location and extent of the lesion. Patients may be asymptomatic, but frequently present with blurry vision. Floaters, photopsias and visual field loss corresponding to the location of the lesion may also occur.1
The patient may experience pain if the lesion compresses ciliary nerves, or from increased intraocular pressure following secondary angle closure. Pain may also arise from iris melanoma seeding into the trabecular meshwork, obstructing aqueous outflow, or from tumor necrosis. However, all of these circumstances are uncommon.1,3,4,12
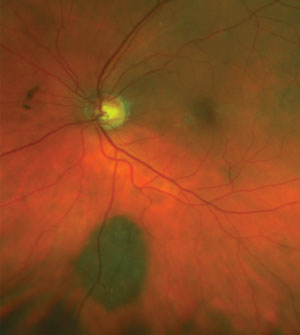 |
| Photo of choroidal nevus, approximately 4mm v. x 3mm h. with overlying drusen. |
Ancillary Testing
While clinical observation is generally adequate for diagnosis, ancillary testing may be helpful.
On B-scan, choroidal melanomas usually appear as solid masses, which exhibit bright anterior acoustic reflection with low internal and basal reflectivity.12,13 On A-scan, low internal reflectivity is seen within the tumor, and oscillation in echo height within the lesion indicates vascularity.12,13 A- and B-scans also aid in accurate tumor size and thickness measurements.
Optical coherence tomography (OCT) and enhanced depth imaging OCT (EDI-OCT) can be quite useful. One study found that choroidal UM usually shows a smooth-domed topography, often with overlying subretinal fluid (SRF) and "shaggy" photoreceptors, which likely represents photoreceptor edema.14 They also show physical compression of the choriocapillaris due to the lesion itself and dark posterior shadowing due to the density of melanin.14,15
Anterior segment OCT imaging can be used for iris melanomas. With this, the surface appears furrowed and the internal reflectivity is variable. However, note that for pigmented iris melanomas, just as with posterior segment OCT, posterior shadowing occurs and the extent of the lesion is often difficult to assess.16
Fundus autofluorescence (FAF) highlights metabolically stressed RPE and, in uveal melanoma, usually shows focal hyperautofluorescence, especially at the borders of the tumor or in areas of SRF or lipofuscin ("orange pigment,"as described below).17 However, the intensity of autofluorescence can vary, and the same pattern may be seen in several other non-UM lesions.18
Fluorescein angiography (FA) does not yield pathognomonic signs for uveal melanoma because findings will vary based on the lesion's pigmentation, vascularity, overlying RPE integrity, etc.12 Thus, FA alone is not very useful in diagnosing UM.
Table 1. Iris Melanoma High Risk Profile
|
On magnetic resonance imaging (MRI), melanomas generally appear hyperintense on T1-weighted images and hypointense on T2-weighted images due to melanin's intrinsic T1 and T2 shortening effects; this does not hold true, however, for amelanotic or lightly pigmented melanomas. MRI is also helpful for determining tumor size and extent of involvement. Ciliary body involvement, extrascleral spread and increasing degree of pigmentation (as gauged by increasing T1 hyperintensity and T2 hypointensity) all carry poorer prognostic implications. Finally, computed tomography (CT) usually shows nonspecific hyperdensity.4
Clinical and FNAB Diagnosis
Most melanomas can be diagnosed clinically. For example, the Collaborative Ocular Melanoma Study (COMS) found that more than 99.5% of large choroidal melanomas were correctly diagnosed clinically.19
For smaller or atypical melanomas, or if the tumor type is less clinically obvious, cytopathology of samples obtained via fine-needle aspiration biopsy (FNAB) will differentiate the tumor with up to 95% accuracy.11,19 Diagnostic FNAB is the most reliable means of differentiating atypical fundus lesions; however, it is only required in approximately 1% to 2% of cases of presumed UM and is usually done at the time of tumor treatment.19-21 FNAB samples may be obtained via trans-corneal, trans-scleral or trans-vitreal approaches. Trans-scleral and trans-vitreal FNAB is generally preferred for pre- and post-equatorial tumors, respectively.11,19 Histology of the solid mass may also be evaluated in enucleated eyes, and excellent correlation, at or near 100%, has been shown between histologic and cytological diagnosis.19
Classification and Prognostics
Uveal melanomas are divided into three histological types: spindle type A, spindle type B, and epithelioid type. Within these histological types, there are four classifications as described by Callender's modified classification:1
| Table 2. Melanoma Mnemonic | |
| Characteristic Thickness Fluid Symptoms Orange Pigment Margin Ultrasound Halo Drusen |
High Risk >2mm Any Any Any Within 3mm of disc Hollow Absent Absent |
- Spindle type tumors
- Epitheloid cell
- Mixed (spindle and epithelia)
- Necrotic
Most uveal melanomas are of the spindle (35% to 45%) or mixed cell types (45% to 63%). Purely epithelioid cell-type melanomas are rare (1.75% to 5%), and necrotic melanomas, for which the predominant cell type is indiscernible, are also rare (5% or less).1,19 Histologically, epithelioid cell categorization and high mitotic count are among the features most predictive of melanoma-related mortality.2,22
The American Joint Committee on Cancer (AJCC) has established an anatomic tumor classification system for anterior and posterior uveal melanomas. Iris melanomas are staged T1-T4 based on amount of extra-iris extension. Posterior melanomas are classified on a T1-T4 scaling criterion based on increasing size, basal diameter and thickness; they are further subcategorized as stage I-IV depending on ciliary body involvement and extraocular extension.5,9
Many techniques are used in molecular testing for chromosomal alterations of UM samples, which are obtained post-enucleation, post-local resection or via FNAB.2 Analysis of chromosomal information from uveal melanomas has identified critical mutations: aberrations in chromosomes 3 (complete or partial loss), 8q (gains) and, to a lesser extent, loss of both 1p and 6q. Polysomy 6p (gains) and disomy 3 are generally associated with longer survival rates for uveal melanoma, while monosomy 3 is associated with poor prognosis.2,23 These gross aberrations are not seen in conjunctival melanoma.2 GNAQ, GNA11, BAP1 and other gene mutations are commonly found, and these genetic markers correlate with increased mortality, particularly in combination with monosomy 3.2,20,23
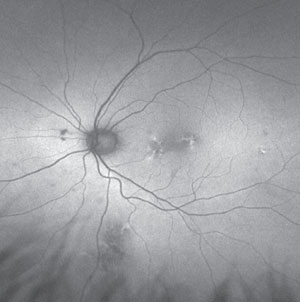 |
| Fundus autofluorescence of choroidal nevus showing overlying patchy hyperautofluorescence. |
Gene expression profiling (GEP) of tumor RNA is helpful for prognostication in primary uveal melanomas. A commercially available assay, Decision Dx-UM (Castle Biosciences), classifies the UM into two metastatic risk classes:
| • | Class 1, with 1B subtype, which classifies UM as low risk and intermediate-risk, respectively. |
| • | Class 2, UM with high risk.2,21 |
At eight years, the survival rate for class 1 is 95%, whereas it is only 31% for class 2.5
Chromosomal analysis and GEP can produce accurate metastasis risk assessments (75% to 95% accurately stratified risk), but the most predictive genetic factor appears to be complete or partial loss of chromosome 3. Those with a normal chromosomal status have a higher rate of survival.2,23 Assessing chromosomal status, in addition to considering AJCC staging offers the best melanoma-related mortality prediction.23
Risk Factors for Formation
There are several identified risk factors for the development of UM. According to one study, the most important risk factors for UM development are pre-existing choroidal nevi and ocular- or oculodermal melanocytosis. Melanocytosis, also known as "nevus of Ota," manifests as grayish pigmentation in the skin, sclera and uvea, and carries a 1/400 lifetime risk of uveal OM development in Caucasians.5 Three percent of patients with UM have ocular melanocytosis.10 Patients with uveal melanoma are at about two to three times higher risk of metastasis if they have ocular or oculodermal melanocytosis.3
Caucasian ethnicity and older age are well-recognized risks.6,9 Additionally, fair skin, poor ability to tan, and those with little ocular pigmentation are at higher risk. A poorer prognosis with UM has also been linked to light iris color, particularly those with light irises but increased choroidal pigmentation.3,6 Choroidal nevi, which are found in 7% of the Caucasian population, are also an important risk factor, as melanomas may develop directly from them. Roughly one in 8,845 choroidal nevi transform into melanomas.6,20
| Iris and Ciliary Body Melanomas Since iris melanomas usually develop from iris nevi, an iris nevus is the most common differential when evaluating for iris melanoma. Remembering the letters "ABCDEF" can help the clinician differentiate between the two. Iris OM is more likely given any of these characteristics (Table 1).3 Ciliary body melanomas may be difficult to detect, as they may remain hidden behind the iris unless they are large, or if they grow through the sclera to produce a visible pigmented episcleral extension. Visible sentinel vessels may form overlying an otherwise hidden tumor, and should prompt further investigation.3 |
Ultraviolet light exposure has long been suspected to play a role in the formation of uveal melanoma. Evidence is mounting that blue light may be a strong risk factor for UM formation, particularly in the setting of pre-existing choroidal nevi.6 For example, arc welding exposure, which is a source of blue ultraviolet light, is significantly associated with UM formation.3,5,6 Prophylactic UV protection is advisable, to help reduce the risks of both OM and a number of other ocular diseases.
Genetic factors such as abnormal alterations in chromosome 3, 6 and 8 and GNAI/11 mutations are other risk factors, as previously noted.6
Differential Diagnosis of Choroidal Melanoma
There are a plethora of differential diagnoses to consider when a lesion looks suspicious for choroidal melanoma.1
• Choroidal nevi. These are present in about 7% of the Caucasian population. Fortunately, <1% of them transform into melanomas, but funduscopically differentiating nevi from a small choroidal UM can be challenging.12,20 OCT findings are often similar to melanomas, but longstanding nevi generally show chronic overlying RPE and outer retinal atrophic changes.14
Clinically, several characteristics of choroidal nevi put them at greater risk for malignant transformation. The mnemonic "TFSOM-UHHD," which stands for "To Find Small Ocular Melanoma—Using Helpful Hints Daily" (Table 2)—may aid in the crucial early detection of UM. The presence of each characteristic raises the risk of malignant transformation from nevus to melanoma threefold, and the presence of three or more of these characteristics is an almost certain indication of melanoma.3,5
Lipofuscin, composed of lipids, proteins and small chromophores, accumulates in the RPE due to metabolic aberration, where the retinal photoreceptor outer segments are incompletely digested, or turn over more quickly than the RPE can phagocytose them. Lipofuscin accumulation may appear as diffusely granular, or form in coalesced areas. This occurrence is better known as "orange pigment," and is best appreciated on FAF, where it brightly fluoresces. The presence of lipofuscin is a prominent risk factor for malignancy—choroidal nevi with orange pigment are highly suspicious. Tumors with SRF also have a higher risk of growth and are more likely to be malignant.17
| Conjunctival Melanoma Conjunctival melanomas account for 5% of ocular melanomas and 2% of all ocular malignant tumors. It is the second most common primary conjunctival malignancy after squamous cell carcinoma. The median age of onset is 60. It occurs in 0.2 to 0.8 per million Caucasians annually; it is seldom reported in non-Caucasians. Usually, conjunctival OM is found in the interpalpebral bulbar conjunctiva, and carries a poorer prognosis if it is elsewhere.2 Differentials include racial melanosis, conjunctival nevi and primary acquired melanosis (PAM).3 Conjunctival melanomas may arise de novo (up to 17%), from pre-existing nevi (up to 25%) or as PAM (up to 76%).2,3 About 10% of PAMs transform to conjunctival melanoma.3 The features of conjunctival OM can vary greatly: from flat to nodular, from amelanotic to darkly pigmented, single to multiple tumors, and the margins may vary; cell type also varies and, like uveal and cutaneous melanomas, may be epitheloid, spindle or a mixture of the two.2 Conjunctival melanomas are classified by the AJCC into T1-T4 based on size and extent of spread. Like UM, increasing rates of metastasis and death occur with increasing class.3 Genetic and histologic outlook for conjunctival OM is not well established.2 Treatment generally involves wide excision with a dry, irrigation-free, "no touch" approach to prevent seeding, followed by topical chemotherapy with mitomycin C, or cryotherapy.2,3 Local recurrence may present in more than 50% of cases.2 Orbital extension necessitates exenteration.3 Metastasis via the lymphatic system develops in 20% to 30% of cases, usually to regional lymph nodes, brain, lung and liver, and is more likely to occur if the tumor has a thickness of >2mm, ulceration, or mitotic counts of >1/mm.2 The 10-year survival rate is approximately 70% to 75%. Frequent monitoring, at least two to three times per year, with lymph node palpation and annual brain MRI and chest x-ray are advised.2 |
Rarely, choroidal melanomas may exhibit a diffuse growth pattern. This diffuse UM is flat with ≤3mm thickness, or <20% ratio of thickness-to-base size. These diffuse melanomas only represent about 3% of uveal melanomas, but they carry a 3.84 times increased risk of metastasis vs. non-diffuse melanoma.3 Diffuse and other small melanomas are quite dangerous, and unfortunately, may easily be misdiagnosed as a benign nevus.
• Choroidal metastatic tumors. Metastatic choroidal tumors (with their primary malignancy elsewhere in the body) tend to be multifocal (averaging two per eye) and often have SRF.11 On average, two out of three primary sites are known, although up to one of out three are not.11 Globe metastases, which originate from primary lung or breast cancer, are usually present in the choroid due to its rich blood supply.4,12 While the lesions may be hard to clinically differentiate from primary uveal melanoma, the symptoms tend to manifest and progress much more quickly.4 EDI-OCT generally shows a bumpier surface topography than primary UM, but like UM, often demonstrates overlying subretinal fluid and shaggy photoreceptors.14
• Melanocytoma. Unlike choroidal melanoma, melanocytomas (also known as hyperpigmented magnocellular nevi) are almost always benign tumors. They are rare and are generally found in patients between 30 and 50 years of age who have dark skin.13
Melanocytomas are darkly pigmented and may occur in the choroid, usually at or near the optic nerve and less frequently in anterior structures such as the sclera, conjunctiva, ciliary body or iris.13,17 They are usually small (<1 disc diameter) and exhibit mild growth in only 10% to 15% of cases. These dense masses are usually avascular and show high internal reflectivity on B-scan, whereas choroidal melanomas are usually more vascular and exhibit low internal reflectivity.
Choroidal fluorescence is generally blocked on FA, because of the heavy pigmentation and lack of vascularity that characterize melanocytomas.13 Also, they are hypofluorescent on FAF due to a lack of lipofuscin, in stark contrast to FAF data in uveal melanoma.17 These lesions also differ histologically from melanomas. When bleached, uniform large cells with a small nucleus, abundant cytoplasm and a clear cytoplasmic membrane are visible. Even with these differences, melanocytomas are still difficult to differentiate from melanomas both clinically and radiologically. B-scan and FA findings can vary in a way that mimic choroidal melanomas, so patients with melanocytomas may, unfortunately, undergo unnecessary treatment for their benign lesion.13
• Disciform macular degeneration. Disciform scarring, subretinal hemorrhaging and the turbid, creamy exudation often seen with age-related macular degeneration (AMD) may cause choroidal elevations and a clinical picture that mimics uveal melanoma. The presence of AMD in the contralateral eye, along with OCT imaging or other ancillary testing, will help differentiate between AMD and uveal melanoma.
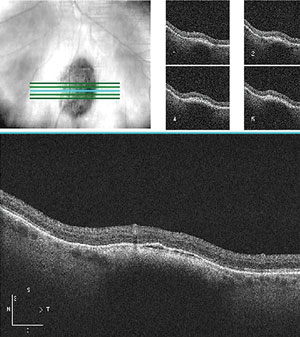 |
| OCT of choroidal nevus showing lesion thickening and thin band of SRF. |
• Peripheral exudative hemorrhagic chorioretinopathy (PEHCR). This retinal degeneration affects elderly patients and appears as an elevated dark mass. On FA, the affected area will show blockage due to hemorrhaging. It appears dense on ultrasound and will usually resolve with time.5,11
• Congenital hypertrophy of the retinal pigment epithelium (CHRPE). These darkly pigmented, flat lesions may be quite variable in size and often have hypo-pigmented lacunae within their borders. They do not grow with time and are benign.
• Choroidal hemangioma. These are benign dilations of the choroidal vasculature and appear as red to orange elevations.10
EDI-OCT reveals smooth, dome-shaped topography and enlargement of the affected choroidal vessel lumen.14 FA indicates a vascular filling pattern and B-scan shows high internal reflectivity.23
Other potentially confusing differential diagnoses include choroidal detachment, granulomas, sclero-choroidal calcifications, vortex vein varix and choroidal osteomas.5
Treatment Options
Several considerations are made when considering treatment for uveal melanoma: tumor size, location, proximity to the fovea and optic nerve, overall systemic health considerations and patient preference.25 The most frequent treatments for posterior UM are enucleation and focal radiotherapy (either proton beam or plaque radiotherapy).3 However, since the COMS revealed that treatment with enucleation or brachytherapy yielded equivalent mortality rates for medium-sized melanomas, globe-sparing procedures are performed when early detection makes them possible.25
• Plaque radiotherapy/brachytherapy. Plaque brachytherapy (otherwise known as plaque radiotherapy) involves placing a radioactive implant or "plaque" into or overlying the cancerous tissue. Iodine-125 and ruthenium-106 isotopes are most commonly used, and around 98% of tumors are locally controlled.3 These plaques are very customizable and can fit almost any ocular site and tumor size (up to 18mm in diameter and 12mm thickness). Some tumors may be technically difficult for plaque placement over their entirety, particularly those that are large or near to the optic nerve.3,20,25
• Charged particle irradiation. Proton beam therapy (PBT) and stereotactic radiosurgery (SRS) are effective for cases of larger, or more inaccessible choroidal melanomas, such as very posterior or juxtapapillary melanomas where plaque radiotherapy may not be possible.
The SRS procedure generally involves retrobulbar anesthesia to immobilize the eye, application of a stereotactic frame, MRI to localize the tumor followed by a large dose of gamma radiation to the lesion.
Tantalum markers (non-metallic clips) are often sutured to the sclera around a posterior lesion to aid in the visualization of the treatment area during the treatment during PBT. Proton beams can then be very precisely focused to deliver radiation to the tumor, largely sparing surrounding tissue. However, the proton beam does deliver radiation to a broader area than SRS. Whereas SRS radiation is usually given in one dose, PBT's larger radiation dose is generally fractioned into multiple sessions.25
PBT demonstrates up to 97% control of local tumors.3 Poorer visual outcomes are generally expected with tumors that are located less than two disc diameters from the fovea or optic disc. One large clinical study comparing SRS and PBT for the treatment of choroidal melanomas found that, for melanomas touching the optic nerve, patients who underwent SRS were more likely to incur severe visual loss. However, PBT and SRS have both proven efficacious and yield comparable rates in regards to tumor control and globe preservation (about 96%).25
Radiation retinopathy is a potential complication of both SRS and PBT; other potential complications include optic neuropathy and neovascular glaucoma.3,25 Secondary enucleation may be necessary, and rates have been reported at 2.4% to 14% post-SRS and 3.7% to 14% post-PBT.25
• Local resection. Uveal melanomas are sometimes locally resected via a procedure that involves excising the entire UM en-bloc—as a full, intact single specimen. Radiotherapy is sometimes used after resection to prevent recurrence.3,25 Tumor seeding is unlikely with local resection, but generally minimal manipulation is attempted.20 This procedure is often used for iris melanomas.
• Enucleation or Exenteration. Advanced melanomas necessitate enucleation. These include tumors >10mm thick, >18mm in basal diameter, those surrounding the optic nerve, or those causing secondary glaucoma. The procedure is done under general anesthesia, and the extraocular muscles are spared to assist with cosmetic prosthetic movement over the volume-replacing implant post operatively. Exenteration of the orbit is usually not necessary unless there are larger degrees of extraocular tumor extension.3
• Transpupillary thermotherapy. TTT uses a diode laser focused through a contact lens, and can treat small choroidal melanomas (<3mm thickness), especially darkly pigmented ones, that do not make contact with the fovea or optic disc.20,25 This modality offers precisely targeted treatment with immediate tumor necrosis, and may be a good selection for elderly patients whose tumor meets the recommended treatment criteria. It can be used as monotherapy, but is now usually done in conjunction with plaque radiotherapy.3,20 Potential side effects include retinal hemorrhaging, vein occlusions or traction, and tumor recurrence in up to 8% of cases.3
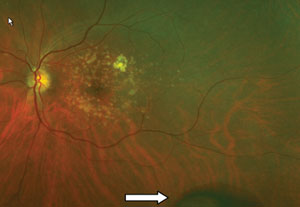 |
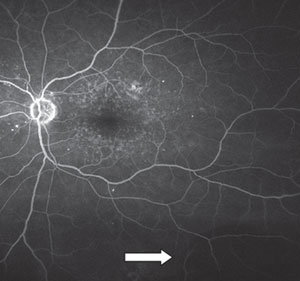 |
| PEHCR and the corresponding fluorescein angiogram, which indicates choroidal blockage due to hemorrhaging. |
Iris melanomas are generally surgically resected, but plaque radiotherapy is often employed, especially if there is a lot of tumor seeding; enucleation may also be necessary.3,4
Observation, while not advised in most cases, may be a reasonable management plan in some patients—the very elderly, frail or those with serious comorbidities may opt out of treatment.
Prognosis
Iris uveal melanoma, the least common of all UM, is relatively non-aggressive and has the best prognosis regarding mortality, especially for younger patients.3,10,26 On the contrary, ciliary body melanomas have the worst prognosis. As previously noted, UM in older patients, larger UM size, diffuse melanoma, extraocular extension, epithelioid cell type, and chromosomal mutations all carry poorer prognosis. Despite tumor size and other risk factors, genetic analysis of the tumor carries significant weight in regards to obtaining an accurate prognosis.2,3,22 Again, complete or partial loss of chromosome 3 is most prognostic for mortality.
Overall, prognosis for UM is poor, with a five-year mortality rate around 40%, and an eventual 50% 10-year mortality rate, primarily due to aggressive metastasis.6,23 Choroidal melanomas metastasize hematogenously (via bloodstream), as the choroid does not have a developed lymphatic system.1 The liver is the most frequent site of metastasis; up to 90% of patients with metastasis have liver involvement.1,4 Prognosis is very poor once liver metastasis is detected, with a median survival duration of only 2.2 to 12.5 months.6 Other metastatic sites include lung, skin and bone.3,4 Molecular genetics are used for prognostication regarding risk of metastatic disease, but, unfortunately, there is little effective treatment for metastatic UM.2
The rate of metastasis and death in uveal melanoma increases with respect to T1-T4 staging criteria by two-, four- and eight-fold, respectively.3,5 Though the metastatic risk is much lower in small melanomas, even small choroidal melanomas do pose a metastatic risk. On average, metastasis is presumed to occur when the UM has a basal diameter of 3mm and a thickness of 1.5mm—at a time when it may be assumed to be a choroidal nevus.5,22 This, again, emphasizes how key early detection is for uveal melanoma. Each millimeter of increase in tumor thickness increases the metastatic risk by 5%.5
Generally speaking, oncologic physicals twice per year with liver function tests, and annual liver imaging (MRI or ultrasound) and chest x-ray are advised to monitor for metastasis.3
Early identification of UM is key, especially given that there is a harrowing difference in metastatic prognosis based on tumor thickness, so differentiating them from other mimickers may save a patient's life. Evolving diagnostic testing not only helps to determine metastatic potential, but in the future may offer more individualized, targeted management strategies to improve survival.
Dr. Weidmayer practices at the VA Ann Arbor Healthcare System in Ann Arbor, Mich. She is also a clinical instructor for the University of Michigan Department of Ophthalmology and Visual Sciences.
References
- Shukla S, Acharya S, Dulan M. Choroidal OM– A Rare Case Report. J Clinical and Diagnostic Research. 2015; 9(5):ED09-10.
- Kalirai H, Coupland SE. An update on ocular melanoma. Diag Histopathol. 21;1:19-25.
- Shields CL, Kels JG, Shields JA. OM of the eye: Revealing hidden secrets, one at a time. Clinics in Dermatol. 2015; 33:183-196.
- Tailor TD, gupta D, Dalley RW, et al. Orbital neoplasms in adults: Clinical, radiologic, and pathologic review. RadioGraphics. 2013;33:1739-58.
- Shields CL,Manalac J, Das C, et al. Choroidal melanoma: clinical features, classification, and top 10 pseudomelanomas. Curr Opin Ophthalmol. 2014;25:177-185.
- Logan P, Bernabeu M, Ferreira A, et al. Evidence for the role of blue light in the development of uveal melanoma. J Ophthalmol. 2015;1-7.
- Shields CL, Shields JA, Materin M, et al. Iris melanoma. Ophthalmol. 2001;108:172-178.
- Shields CL, Shields PW, Manalac J, et al. Review of cystic and solid tumors of the iris. Oman J Ophthalmol. 2013; 6(3):159-164.
- Shields CL, Kaliki S, Furuta M, et al. American joint committee on cancer classification of uveal OM (anatomic stage) predicts prognosis in 7731 patients. Ophthalmol. 2015;122:1180-1186.
- Sivalingam MD, Hasanreisoglu M, Shields CL. Choroidal OM in children: be aware of risks. J Pediatr Ophthalmol Strabismus. 2014;51:e85.
- Aziz HA, Martel JN, Viscotti CV, et al. This, that or something different? Survey Ophthalmol. 2015; np Web. 21 May 2015. 27 July 2015.
- Weidmayer SL. Choroidal OMis a Life Sentence. Rev Optom. 2012;46-55.
- Ahmad SS, Lad L, Ghani SA. A case of choroidal melanocytoma mimicking a choroidal melanoma. Saudi J Ophthalmol. 2015;29:242-45.
- Shields CA, Manalac J, Das C, et al. Review of spectral domain enhanced depth imaging optical coherence tomography of tumors of the choroid. Indian J Ophthalmol. 2015;63(2):117-21.
- Dunbar M. Significant Growth Potential? Rev Optom 2014;93-94.
- Williams C, Page EM. Technology is key to iris lesion diagnosis. Rev Optom. 2009. Available at www.reviewofoptometry.com/contant/d/case_report/i798/c/14849/.
- Salvanos P, Utheim TP, Moe MC, et al. Autofluorescece imaging in the differential diagnosis of optic disc melanocytoma. Acta Ophthalmol. 2015; 93:476-480.
- Reznicek L, Stumpf C, Seidensticker F, et al. Role of wide-field autofluorescence imaging and scanning laser ophthalmoscopy in differentiation of choroidal pigmented lesions. Int J Ophthalmol. 2014; 7(4):697-703.
- Medina CA, Biscotti CV, Singh N, Dingh AD. Diagnostic cytologic features of uveal melanoma. Ophthalmol. 2015;122:1580-1584.
- Shields JA, Shields CL. Management of posterior uveal melanoma: past, present, and future. Ophthalmol. 2015; 122:414-428.
- Klufas MA, Itty S, McCannel CA, et al. Variable results for uveal melanoma—specific gene expression profile prognostic test in choroidal Metastasis.
- Malclès A, et al. Small metastasizing choroidal melanomas. Acta Ophthalmol. 2015;93:e160-66.
- Bagger M, Andersen MT, Andersen KK, et al. The prognostic effect of american joint committee on cancer staging and genetic status in patients with choroidal and ciliary body melanoma. Invest Ophthalmol Vis Sci. 2015;56:438-44.
- Rafieetary M, Huddleston S, Sigler E. differential diagnosis of retinal disease. Rev Optom. 2015;34-42.
- Sikuade MJ, Salvi S, Rundle PA, et al. Outcomes of treatment with stereotactic radiosurgery or proton beam therapy for choroidal melanoma. Eye. 2015;1-5. [Epub] 26. Damato BE, Heimann H, Kalirai H, Coupland SE. Age, survival predictors, and metastatic death in patients with choroidal melanoma: tentative evidence of a therapeutic effect on survival. JAMA Ophthalmol. 2014;132(5):605-613.