Identifying Systemic Sources of Uveitis
By Aaron Bronner, OD
Release Date:
November 2016
Expiration Date:
November 15, 2019
Goal Statement:
Uveitis is often associated with serious ocular and systemic conditions requiring immediate treatment. This course provides an overview of the most common etiologies underpinning uveitis in the United States. The various causes of uveitis as well as creating a workable differential diagnosis in the different cases are discussed.
Faculty/Editorial Board:
Aaron Bronner, OD
Credit Statement:
COPE approval for 2 hours of CE credit is 51560-SD for this course. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statement:
The author has no relationships to disclose.
Uveitis is a challenging ocular condition to manage due to its link to a wide variety of systemic inflammatory and infectious processes, as well as its often under-appreciated potential to cause vision loss. Approximately 40% to 50% of all uveitis cases are caused by an underlying systemic condition. We recognize some of the clinical variability among etiologies, but our findings don’t always mean what we think. While lab studies are sometimes critical for diagnosis, ordering a huge battery of tests is neither cost effective nor particularly useful.
 |
| Cell in the anterior chamber is the diagnostic finding of anterior uveitis. Click image to enlarge. |
This article reviews the signposts we can use to whittle our differential diagnosis down to a smaller set of conditions prior to testing. Because anterior uveitis accounts for 70% of cases, it is the primary focus of this review.
Clinical Clues
First, consider factors in the presentation and patient demographics that might allow you to narrow your focus.
Certain underlying etiologies have very well described propensities for manifesting in certain ways—granulomatous forms of uveitis generating mutton fat keratic precipitates (KPs), for example. However, these associations are not set in stone; HLA-B27, in rare cases, may cause mutton fat KPs, while sarcoidosis-associated uveitis does not present with mutton fat KP or iris nodules—classic signs of granulomatous disease—in many cases.2,3 Additionally, recognizing trends in KP distribution can be enormously beneficial. KPs classically present in Arlt’s triangle, while diffuse, very localized or linear KP distributions are strongly suggestive of specific etiologies.4,5
Although we tend to think of uveitis symptomology in terms of photophobia and a deep achy eye, this is only characteristic of acute anterior uveitis (AAU). Patients with deeper inflammation, such as primary vitritis, choroiditis or chronic anterior disease, do not experience these classic symptoms; rather, they complain of reduced vision or floaters.5 These groups all have separate etiologies with only minimal spill over. For this reason, asymptomatic uveitis patients should generate a separate differential than those with classic symptoms.
Given the wide array of systemic conditions associated with uveitis, identifying available clues can help you more easily determine the cause. More specifically, looking at the intersection of both the clinical picture of the episode and the patient can give you a good start towards a diagnosis. For example, one study on tubulointerstitial nephritis and uveitis syndrome (TINU), demonstrates that 1.7% of the patients presenting to this uveitis center had TINU, making it a rare source of disease.6 However, when the authors looked at only uveitis cases that were acute, bilateral and anterior and occurred in patients younger than 20, they found that TINU accounted for 32% of all cases.6 By simply assessing an obvious patient feature (age) and clinical features (acute vs. chronic and laterality), the researchers increased the likelihood they were dealing with TINU by 19 times, making this rare disease the primary differential in the correct setting.6
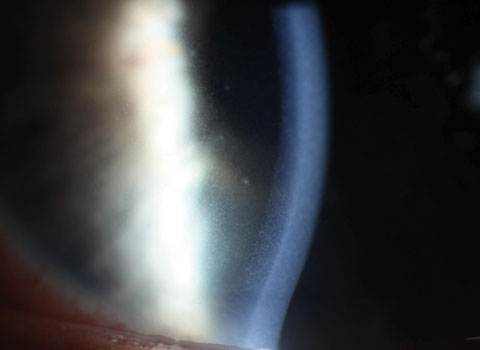 |
| Fine KP dusting of the endothelium was seen in a case of HLA-B27 AAU. This is frequently encountered in cases of nongranulomatous anterior uveitis. Click image to enlarge. |
While not all sources of uveitis are as easily differentiated, demographic factors such as age and race play important roles in determining likely etiologies. And, be aware that the etiologies presented here are common to the United States, which vary in frequency across the globe.
Etiologies
Much like “dry eye” is a catch-all phrase for a heterogeneous assortment of often unrelated conditions, so too is “uveitis” an umbrella term with poor specificity. Below we review particular etiologies with an emphasis on how to recognize their unique attributes.
• HLA-B27. Specific human leukocyte antigens (HLAs)—cellular markers associated with the immune response—are associated with an increased risk of specific disease processes. HLA-B27 is associated with an increased risk for developing diseases such as ankylosing spondylitis, reactive arthritis, psoriatic arthritis and inflammatory bowel disease. Additionally, its presence may be associated with local forms of inflammation such as isolated aphthous ulcers (canker sores), erythema nodosum or anterior uveitis.
A person who carries HLA-B27 has a 100-fold greater relative risk for AAU over a population-matched non-carrier of the marker.5 HLA-B27’s association with uveitis accounts for approximately 50% of AAU cases in Caucasian populations, superseding idiopathic causes.5,7,8 This association increases to 70% in patients with recurrent AAU.10 Patients with HLA-B27 and an associated systemic disease have a 25% lifetime risk of developing an episode of AAU, and those with a history of HLA-B27 and AAU have a 50% risk of developing an associated disease.1,5,6
Patient setting. The patient who develops an initial bout of HLA-B27 uveitis will generally be a teen aged to middle aged Caucasian, and one study reports that six out of 138 uveitis patients over the age of 60 developed HLA-B27 uveitis as a primary episode.14 The incidence of HLA-B27 among Caucasians is approximately 8%, and its rate is even higher among certain indigenous American populations such as Haida Native Americans (50%) and American Inuit populations (25%).1 In African Americans, the incidence drops to 2%, and if it exists at all in an unmixed African racial profile, it’s only at extremely low levels.9 While most cases of uveitis across all populations are idiopathic, among Caucasians with AAU, HLA-B27 is the most common etiology.1,5,7
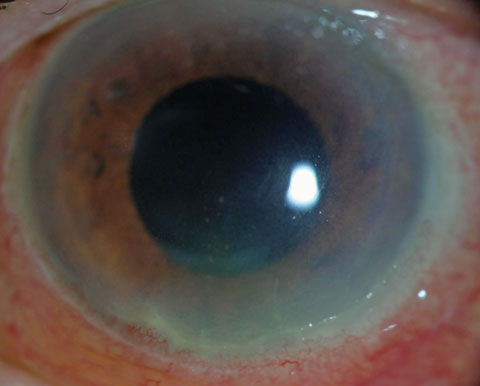 |
| Seen here are early granulomatous KP respecting Arlt’s triangle in a case of sarcoidosis-related uveitis. Arlt’s triangle distribution is a general finding and not suggestive of any specific etiology. Click image to enlarge. |
Clinical picture. On history and exam, a patient with HLA-B27 uveitis will generally look like the classic case of moderate-to-severe unilateral acute, non-granulomatous uveitis. The patient will be symptomatic with photophobia and vague deep discomfort. The exam will show ciliary flush and white cells predominating in the anterior chamber with significant flare as well. Fibrin and hypopyon occur in a high percentage of patients and vitreal spillover is frequently seen, though anterior chamber inflammation is always primary.1,7,10
While cystoid macular edema (CME) and epiretinal membrane (ERM) may develop, especially with recurrences, the prognosis of HLA-B27 uveitis is generally good if treated early, with only idiopathic disease having better outcomes.5 Each attack lasts for an average of six to eight weeks, and treatment should continue over that time frame. Recurrences may freely alternate between eyes, and their timing varies dramatically, with the average recurrence being every one to two years.1
• Juvenile idiopathic arthritis (JIA). Although the precise mechanism of JIA is not known, patients can generally be subclassified based on joint involvement and serologic testing. The condition exhibits significant heterogeneity, so several subtypes are possible, which include the pauciarticular (fewer than five joints involved within the first six months), polyarticular (five or more joints involved within the first six months) and the systemic form, which is paired with features such as fever, rash and internal organ involvement.
The pauci- and polyarticular forms may be further subtyped as rheumatoid factor positive or negative and anti-nuclear antibody (ANA) positive or negative, with ANA(+) pauciarticular patients having a 30% risk of uveitis.5,10,11 Conversely, patients with rheumatoid factor-associated disease or systemic JIA have a low risk of developing uveitis.10
Finally, a JIA cohort exists that is predominated by older male patients who develop a disease form similar to HLA-B27 uveitis, and may go on to develop associated spondyloarthropathies.10-12
Patient setting. Very young patients with asymptomatic uveitis tip the clinician off to JIA. These patients most typically develop arthritis between the ages of three and six; when uveitis develops, it generally follows within the year, though in approximately 5% of cases the uveitis precedes the arthritis.12 Uveitis in teenage males—a subgroup of JIA-associated patients—tends to be more acute and symptomatic. This group is often associated with HLA-B27 and may develop an associated adult form of spondyloarthropathies.10
 |
| The fibrin pupillary plaque seen here resulted from HLA-B27 uveitis. The fibrin in the anterior chamber is common in this etiology. Click image to enlarge. |
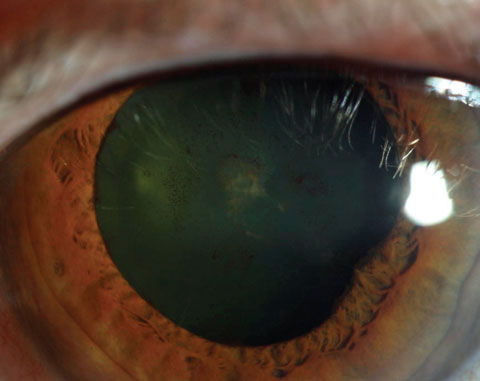 |
| Resolution of the plaque should occur after episode has been controlled, but cataract may result. Click image to enlarge. |
In any juvenile patient with white blood cells within the eye, particularly those that are asymptomatic, keep the masquerade syndromes (discussed below) such as leukemia on the differential list.
Clinical picture. In JIA uveitis, the inflammation is confined to the anterior segment, most typically bilateral, mild and chronic in nature. In contrast to the significant symptomology seen in HLA-B27-associated disease, JIA patients are asymptomatic in most cases.12 The eyes are often white, which may belie an anterior chamber reaction that varies from very mild to severe.5,12 KPs reflect the nongranulomatous nature of the disease and are fine or medium sized. Because the uveitis generally follows the arthritis, a systemic diagnosis of JIA is often pre-existing.
As with all chronic forms of uveitis, potential sources of vision loss in JIA patients include glaucoma, band keratopathy and cataract, with the latter developing in approximately 25% of eyes with JIA.11 Due to the link to sight-threatening conditions and the fact these patients will often be asymptomatic, ANA(+) JIA patients will require follow up every three to four months and ANA(-) patients every six months, even in the absence of diagnosed uveitis, to rule out its development. Patients with the systemic subtype may be followed yearly to ensure no insidious causes of vision loss are developing.
The older male subtype of JIA is more in line with seronegative spondyloarthropathies in adulthood than younger cohorts, and this uveitis behaves more like that associated with HLA-B27.
• Viral uveitis. While viral causes such as herpes simplex virus (HSV) 1 and 2 and varicella zoster virus (VZV) can cause pathology in any age group, VZV especially tends to be more prevalent in patients older than 60 years, while newly acquired autoimmune conditions tend to decrease beyond the age of 60. Therefore, we should be increasingly suspect about infectious etiologies of uveitis, particularly those etiologies which affect an aging population, in an elderly patient with new uveitis. Herpes zoster and herpes simplex do account for a higher percentage of uveitis cases as we age, with one retrospective review showing viral sources of uveitis as being the causative etiology in 5% of their cases overall, but approximately 18% of cases in patients older than 60.14
Patient setting. In patients over the age of 60 with a first episode of anterior uveitis and no other risk factors (particularly for masquerade syndromes—see below), think “viral uveitis” until proven otherwise. As viral forms of uveitis are often paired with corneal edema, patients with herpetic uveitis will frequently have a chief complaint of blurred vision.
Clinical picture. While almost exclusively unilateral, viral uveitis may be either granulomatous or nongranulomatous; reports are inconsistent as to which form predominates.4,5 In HSV, acute, recurrent episodes are the norm, while VZV uveitis tends to be more chronic. Most typically, it manifests as a keratouveitis, with keratic precipitates and concomitant corneal edema, both in a circular distribution in the central cornea. However, alternate forms with diffuse and linear KPs are also encountered and are strong indicators of a herpetic etiology. Diffuse KPs are only typical of viral uveitis, toxoplasmosis and Fuchs’ heterochomic iridocyclitis.5 It can be encountered in the absence of corneal involvement, in which case deeper clues to the diagnosis may be found.15 IOP is elevated in up to 90% of cases.16 Sectoral iris atrophy occurs most classically in VZV uveitis, but is also found in HSV cases.4,5,16 Posterior synechiae are less common in viral uveitis than other forms of AAU.4 Panuveitis as either progressive outer retinal necrosis or acute retinal necrosis is rare, but mandates immediate retinal referral.
Most herpetic uveitis cases can be diagnosed clinically based on the associated findings and patient demographics with good reliability, though PCR of aqueous offers a slightly invasive option to confirm diagnosis.
 |
| Above, this diffuse distribution of KP was seen in a case of Fuchs' heterochromic iridocyclitis (FHI). This KP distribution is diagnostically suggestive of herpetic disease, FHI and anterior spill-over of toxoplasmosis, and is also seen in corneal allograft rejection. Below, linear endotheliitis was seen in a case of viral keratouveitis—the dendritic pattern seen is actually a KP pattern on the endothelium. This is also encountered in corneal allograft rejection. Click images to enlarge. |
 |
• Sarcoidosis. This is a multisystem disorder of unknown etiology, though an infectious trigger is widely suspected, which results in the development of non-necrotizing granuloma formation throughout the body.17 Classically, the lungs are the most often involved organ, and ocular involvement is close behind with 25% to 60% of cases affecting the eyes; however, most patients with sarcoid will be asymptomatic.17,18,20 Systemic involvement, and the precise degree of ocular involvement, varies among age groups and ethnic profiles.3
While uveitis is only one of the many ocular manifestations of sarcoid, it is also the most common. Ocular involvement may occur in the absence of systemic involvement (which is nonspecific), and pulmonary findings may wax and wane throughout the course of the disease, making it a difficult disease to diagnose.20 Because of this, some epidemiologic studies and classification systems will list both confirmed and suspected sarcoidosis as causes of uveitis, and at least one expert suspects undiagnosed sarcoid accounts for a significant percentage of idiopathic uveitis.14,18,20 In a large retrospective review on chronic uveitis etiologies, sarcoid was the second most common after idiopathic at 14%.19 This figure only takes into account sarcoid presenting as the more common chronic form, but sarcoid also appears less frequently as a source of acute disease.
Patient setting. Sarcoidosis can affect any population, though women are affected more frequently than men. In the United States, African Americans develop sarcoid 10 to 17 times more than Caucasians. Although sarcoid has become the chief source of ocular inflammation in Japan, North American patients of Asian ancestry have a lower risk than those living in Japan.3,5,18,21
Age of incidence varies depending on race. Caucasian patients are more likely to have sarcoid uveitis develop after the age of 50, while African Americans tend to develop it roughly a decade earlier.3,21
In juvenile-onset sarcoidosis, the disease presents with more arthritis symptoms and is on the differential for any childhood arthritis or uveitis.13
Clinical picture. Sarcoid is often an asymptomatic disease, and when symptoms occur they are most often nonspecific such as a dry cough or fatigue.20 In 80% of cases, sarcoid uveitis presents bilaterally.18,19 While sarcoid is the chief source of granulomatous uveitis and may present with mutton fat KPs, iris nodules or both, it frequently presents without either of these key markers.3,21 Iris nodules, when present, may occur at the pupillary margin (known as Koeppe’s nodules) or within the stroma of the iris (busacca nodules). Berlin’s nodules, a less frequently recognized form of anterior segment nodules associated with granulomatous disease, occur at the trabecular meshwork (TM). These can be difficult to detect as they are often white and very small.18 Tent-shaped peripheral anterior synechiae (PAS) in an eye with concomitant uveitis is also suggestive of sarcoid.18
Despite sarcoid being a granulomatous disease, granulomatous findings are not always present. For instance, in one study, 66% of patients with sarcoid-linked uveitis had nongranulomatous anterior uveitis on presentation.21
Note that anterior uveitis is only one of sarcoid’s uveal manifestations. It also causes intermediate uveitis, and is the chief systemic differential for patients presenting with primary vitritis, which manifests as white cells in the anterior vitreous and accumulation of acellular inflammatory debris (i.e., “snowballs”) in the inferior vitreoretinal interface—known as snowbanking—the latter of which is quite suggestive of sarcoid.18 Retinal vasculitis, primarily exudative periphlebitis (inflammation of retinal venules), is a classic sign of sarcoid, a finding often described as candlewax drippings.18 In severe cases, exudative periphlebitis may culminate in a retinal vein occlusion.5,18 Choroiditis may manifest as solitary choroidal nodules or smaller, multifocal-type choroiditis. These small, yellow choroidal lesions may resolve completely or result in small, punched-out scars, similar to those seen in histoplasmosis.5,18,20
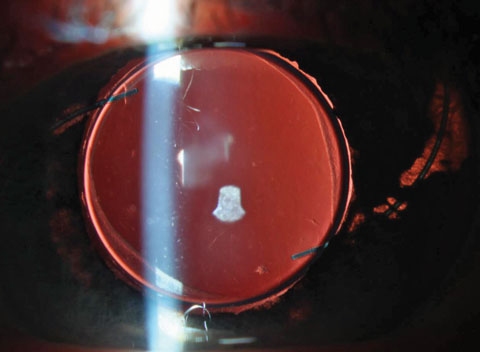 |
| This uveitis masquerader was caused by obvious mechanical chafing of IOL haptics. These may go on to cause micro-hyphema and elevated IOP at which point it would be classified as uveitis glaucoma hyphema (or UGH) syndrome. Click image to enlarge. |
Sarcoid uveitis may present with any or all of these findings. However, racial differences influence which are found. African Americans may have a significantly higher rate of anterior segment granulomatous disease when compared with Caucasians, with research showing 72% of African Americans with sarcoidosis and uveitis exhibit anterior granulomatous findings compared with 25% of Caucasians.3 Since a significant percentage of cases occur in the absence of its classical presentation, awareness of the alternate, deeper forms of sarcoid uveitis are very important for clinical decision making.
Prognosis for sarcoid uveitis follows, to a degree, the depth of uveal involvement. Patients who develop CME or multifocal choroiditis are at the greatest risk for vision loss, and patients with anterior only disease have a relatively good visual prognosis.5,22,23
• Syphilis. This is no longer a primary source of uveitis, with most estimates of luetic disease accounting for 0.5% to 3% of all uveitis cases.25,26 Still, its varied presentation and link to severe ocular and systemic morbidity keeps it on the differential list in any adult patient.20,23,26-28 Ocular manifestations of syphilis are extremely varied, and syphilis serologies should be part of essentially any systemic inflammatory workup. Though ocular involvement is uncommon, uveitis is the most frequent ocular manifestation and may be found in 2.5% to 5% of all cases of tertiary disease.23,25 In its acquired form, luetic uveitis may occur in the disease’s primary stage but is more frequently encountered during secondary or latent stages and may also be associated with neurosyphilis.5,23,27,28 Coinfection with HIV may cause altered pathologic patterns of the disease, including its uveal manifestations.25,27
Patient setting. Of the approximately 36 million people worldwide living with syphilis, 90% are in developing countries.28 In the United States, men account for roughly 85% of all cases of acquired syphilis.24,28 A spike in infections during the 2000s was driven primarily by male-male intercourse.24,28 This group accounted for 64% of cases of patients with primary or secondary syphilis in 2004.23 The disease has also disproportionately affected African Americans, with a rate of 9.0/100,000 (compared with the national rate of 2.7/100,000).24
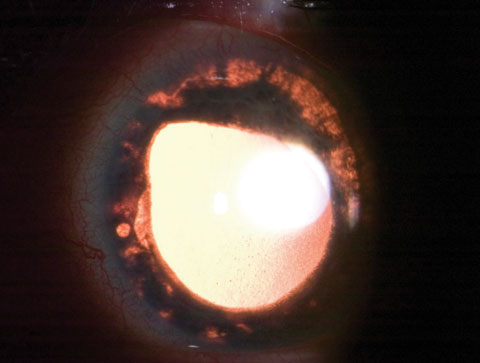 |
| This sectoral iris atrophy was seen in a patient with zoster iritis. Click image to enlarge. |
• Tuberculosis (TB). Worldwide, nearly two billion people are infected with Mycobacterium tuberculosis, of which 10% develop frank tuberculosis.30 The vast majority of these cases occur in developing countries where poverty is a primary risk factor.31 TB remains the primary source of infectious morbidity and mortality across the globe.31,32
As the organism is an obligate aerobe, it most commonly affects parts of the body with high oxygen concentrations, such as the lungs.31 Not surprisingly, uveitis is the most common ocular manifestation of TB, as the highly vascularized uveal structures (particularly the choroid) are among the most highly oxygenated tissues of the body.31 Despite this, in the United States, TB accounts for only 0.5% of cases at uveitis clinics.32
Patient setting. History-based risk factors are essential and are advocated as part of clinical criteria for diagnosing ocular tuberculosis. Immune suppression is the primary risk factor for TB in the United States and developed countries, though emigration from endemic areas or previous exposure to those with active pulmonary TB are equally important.31,32 While pulmonary symptoms may help steer clinical suspicion, general symptoms such as fever, night sweats or fatigue may be of some help in the absence of pulmonary findings.31
Using Lab Tests to Hone the DifferentialLabs are a critical component of uveitis management. The following is a brief primer on a handful of labs commonly used to manage anterior uveitis. • HLA-B27. A positive result (which is 99% sensitive and specific for the antigen) indicates the presence of one of the HLA-B alleles. The test itself is not diagnostic for any specific disease; however, a positive result in the presence of unilateral acute anterior uveitis is diagnostic. Patients with a positive HLA-B27 test without a previously diagnosed systemic disease should be counseled appropriately on their systemic risks.42 • Antinuclear antibodies. Most often, ODs think of ANA as a test for lupus. However, these are associated with a variety of systemic diseases and even exist in the normal population. While ANA is associated with lupus, isolated uveitis is uncommon in systemic lupus erythematosus (SLE). ANA testing for SLE (a rare cause of uveitis) has a low positive predictive value and should not be tested for on a routine basis.43,44 ANA’s value in uveitis management is part of a broad systemic workup or when subclassifying JIA. • Rheumatoid factor (Rf). Like ANA, Rf’s utility in uveitis screening should be, in most cases, reserved for classification of JIA. Rheumatoid arthritis seldom produces isolated uveitis. • Angiotensin converting enzyme (ACE). This is a byproduct of macrophages interacting with granulomas.20 Elevated serum ACE, while associated with sarcoidosis, is not specific for the disease. It’s also not particularly sensitive for sarcoid, with only 40% to 60% of sarcoid patients having elevated levels.3,18 The best testing for sarcoidosis combines serum ACE testing and a chest X-ray or CT. ACE inhibitors limit the reliability ACE testing.18 • Chest X-rays. These are most frequently used to test for sarcoid and tuberculosis. When used for sarcoidosis, bilateral hilar lymphadenopathy (BHL) is the significant finding. As with ACE testing, chest X-ray is not highly sensitive for the sarcoid with only 50% to 80% being positive.3 Further, it is possible for the chest X-rays to alternately be positive or negative throughout the course of the disease.20 When combined with serum testing, however, sensitivity of the two tests together rises to 89%.3 BHL may also occur in lymphoma.18 Though chest X-rays will be more frequently ordered for sarcoid, lobe cavitation is the key finding when used for TB, though less than half of TB patients with ocular symptoms will have pulmonary involvement.30 • Syphilis testing. The syphilis organism Treponema pallidum can’t be cultured and is difficult to see under light microscopy, so testing relies on the body’s reaction to elements associated with the bacterium. Testing can be broken down into to two general types: Non-treponemal tests: Venereal Disease Research Laboratory (VDRL) and rapid plasma reagin (RPR). Treponemal tests: Fluorescent treponemal antibody absorption (FTA-ABS) and micro-hemagglutination for Treponema pallidum (MHA-TP). The non-treponemal tests are less expensive and less specific; they are often negative during primary and latent syphilis, though will also be negative after successful treatment. Treponemal tests, which look for specific antibodies to the organism, are more sensitive and will be positive during primary syphilis and latent syphilis. They will also remain positive after treatment.24 • Tuberculosis skin prick. This uses a type IV hypersensitivity reaction to Mycobacterium tuberculosis (TB) antigens. Infected patients develop a large area of induration (swollen, hardened skin). As expected, patients who are immunosuppressed will have a less vigorous zone of induration and may generate false negatives. Patients from countries where the Bacillus Calmette-Guerin (BCG) immunization is given for TB may also have false negatives.34 Since TB is an uncommon source of uveitis, routine testing is unnecessary unless history and exam are supportive. |
Clinical picture. Posterior and panuveitis are the most common forms of TB uveitis.32 Serpiginous choroiditis, retinal vasculitis with or without choroiditis and broad-based posterior synechiae are suggestive of the disease, being quite specific but only weakly sensitive for TB.30 Their presence strongly merits evaluation for TB in an at-risk population, but their absence does not rule out the condition. In its anterior form, TB most commonly causes chronic recurrent uveitis characterized.
While TB is a granulomatous process, mutton fat KPs or iris nodules do not occur much more than in a non-TB uveitis population.30 Though TB is not on the primary differential for most episodes of uveitis, its presence should be considered prior to initiating any treatments that rely upon a mechanism of systemic immune suppression.
• Fuchs’ heterochromic iridocyclitis (FHI). Proposed mechanisms of FHI include infectious sources (toxoplasmosis, HSV or VZV), disorders of adrenergic innervation, genetic and autoimmunity to anterior segment self-antigens.35 More recently, researchers have made a very compelling case for a rubella etiology, by showing that patients with FHI demonstrate a universal immunoglobin activity against rubella.5,36,37 A high percentage of Fuchs’ patients have rubella RNA recovered from their anterior chamber, and FHI incidence is lower in countries with rubella vaccination programs compared to countries without programs.5,32,37
Patient setting. While FHI may occur in any demographic, it’s most commonly seen in a young and middle-aged adult population.33,34 It affects Caucasian and African American populations, though heterochromia is less prominent in deeply pigmented populations, possibly leading to underreporting in the African American population.38
Clinical picture. The vast majority of cases are unilateral and chronic with mild anterior chamber reaction and a white eye. Despite being a frequently missed diagnosis, FHI is not rare; one study reports it as the second most common etiology of chronic uveitis behind sarcoidosis, accounting for 12% of chronic cases at one uveitis clinic over 35 years.19 Patients are nearly always asymptomatic, and researchers suggest that, even prior to evaluation, the picture of a young, healthy adult with asymptomatic uveitis and good vision should alert the clinician to FHI as a possibility.33
FHI is associated with iris heterochromia. In a patient with light eyes, the darker iris is involved; in dark eyes, the lighter iris is involved. While heterochromia is an important—nearly pathognomonic—finding, as many as 30% of FHI cases lack this feature.34
Diffuse, white stellate KPs may be found in up to 100% of FHI patients.5,34 Less common are iris nodules, which occur more frequently in African American FHI populations; posterior synechiae are almost always absent.34,38 Vitritis is also frequently encountered, though it does not produce snowbanking as seen in pars planitis or other forms of intermediate uveitis, and retinal scars are often seen.33,34
Later in the course of the disease, cataracts almost always manifest as mid-lens opacities or posterior subcapsular changes.33,34 Additionally, glaucoma occurs in 9% to 50% of eyes with FHI over time.34 Due to its asymptomatic nature, delays in diagnosis are common, with patients often aware of heterochromia years prior to visual concerns that are linked to cataract or glaucoma.
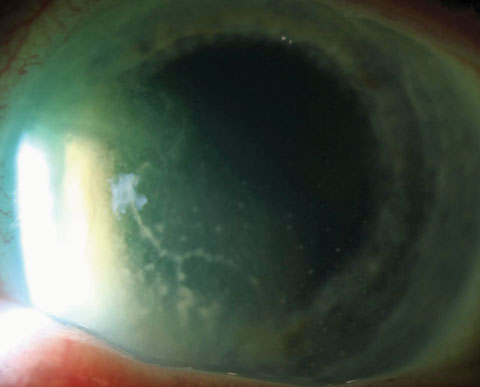
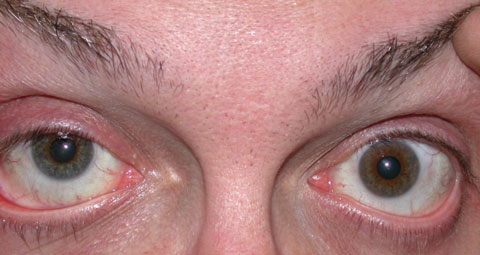 |
| Heterochromia as encountered in FHI is seen here. Heterochromia may not be present or may manifest as more irregular iris depigmentation. |
Given its proposed infectious etiology, FHI does not respond, or responds incompletely, to corticosteroids.33,34 Treatment should focus instead on preventing vision loss from lens and vitreous opacities, as well as the timely diagnosis and management of glaucoma.5,33,34
Masquerade Syndromes
These conditions produce inflammation or pseudoinflammation in the eye, but stem from noninflammatory conditions such as lymphoma, exogenous sources or surgical complications. Among each relevant population, consider the possible masquerade syndromes when formulating a differential diagnosis.
• Intraocular lymphoma. Vitritis is a universal finding in intraocular lymphoma. The form most associated with ocular findings is non-Hodgkin’s CNS lymphoma, which tends to present between ages 50 and 70.5,15,38 Retinal findings are variable and only 20% of cases may have anterior uveitis.15 Any case with chronic vitritis in a patient older than 50 warrants suspicion for lymphoma. Workup includes head MRI, lumbar puncture and diagnostic vitrectomy.5,38
• Intraocular leukemia. Although this masquerades as uveitis less commonly than lymphoma, acute leukemias account for up to 5% of pediatric uveitis cases.39 Because the presence of uveitis is a poor prognostic sign—it usually indicates spread to the CNS or bone marrow relapse—it is enormously important to consider intraocular leukemia as the etiology in this population.39 Hypopyon, pseudohypopyon and increased IOP as a result of leukemic infiltration of the TM are all possible findings. When IOP is sufficiently high, the patient may be symptomatic; otherwise, the patient will be asymptomatic.
• Retained lens material after cataract surgery. In any patient with a recent history of cataract surgery preceding chronic intraocular inflammation, retained lens fragments must be suspected as a primary differential. While uncomplicated cataract surgery and IOL placement almost universally produces white cells in the chamber, this is not an immune reaction in the typical sense. Rather, the mechanical trauma of the surgery disrupts the blood/aqueous barrier, resulting in white cells leaching into the anterior chamber. Under most circumstances, the blood/aqueous barrier is reestablished within two to four weeks, even without corticosteroids, and the chamber becomes quiet. Any lingering inflammation should produce a strong suspicion that lens material, which is an antigenic source of uveitis, may be causative. Thoroughly evaluate the angle with gonioscopy and the posterior chamber with dynamic viewing through a dilated pupil. If a fragment is still not visible, refer for ultrasonic biomicroscopy. Aspiration of any long-lasting lens fragment is necessary.
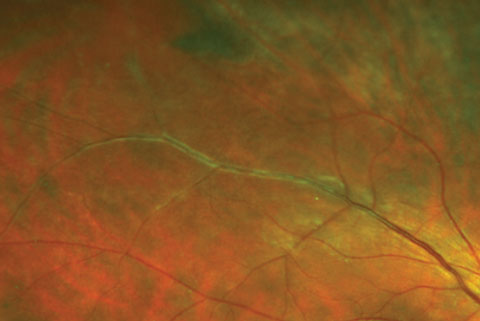 |
| Here, venous sheathing was caused by early periphlebitis as seen in sarcoid uveitis. The vasculitis in these cases may become occlusive and lead to BRVO. Click image to enlarge. |
• Uveitis glaucoma hyphema (UGH) syndrome. This is most common with anterior chamber IOLs, caused when chafing of the IOL on the iris results in a mechanical iritis, hyphema and inflammatory glaucoma. Lenses placed in the ciliary sulcus have a lower risk, though chafing and UGH may still occur. IOLs placed fully in the capsular bag present no risk. Cycloplegics (to reduce chafing associated with pupillary movement), corticosteroids and IOP-reducing medications are palliative, but IOL repositioning is the long-term treatment when feasible.
• Chronic infectious endophthalmitis. Chronic cells in the anterior chamber and vitreous following cataract surgery, where lens fragment and IOL positioning issues have been ruled out, means chronic infectious endophthalmitis should be considered. This masquerade condition produces a milder intraocular inflammation than the acute form. Assessment of the lens capsule for suspicious white plaques can be helpful to steer suspicion. Diagnosis is confirmed through anterior chamber and or vitreous tap and culture.
• Brimonidine side effect. An infrequently encountered though important late side effect of brimonidine use is acute granulomatous uveitis. This occurs approximately one year after initiating therapy and responds to discontinuation. The uveitis will recur if the medication is reintroduced and occurs only in medicated eyes.41
• Retained intraocular foreign body. Iron-based foreign bodies are antigenic and will produce a cellular response. In some cases, patients will be unaware of the injury but will come in with complaints of uveitis weeks or months later. Consider this: if uveitis is not responding permanently to therapy, question patients for a history of welding, grinding or frequent use of a striking instrument. Gonioscopy can help identify material in the angle. The uveitis in these cases will be unilateral, chronic and generally mild. It will respond to steroids temporarily but will recur upon cessation.
Despite the enormous variety in uveitic etiologies, often relatively narrow differentials can be made when clinicians carefully consider the clinical appearance and the patient setting of the disease. By becoming familiar with and assessing these features, the appropriate diagnosis and management of uveitis, though still difficult, can become less opaque and simpler to achieve.
Dr. Bronner is an attending optometrist at Pacific Cataract and Laser Institute in Kennewick, WA.
|
1. Chang JH, McCluskey PJ, Wakefield D. Acute anteriror uvetitis and HLA-B27. Surv Ophthalmol. 2005;50:364-88. 2. Miserocchi E, Modorati G, Di Matteo F, et al. Visual outcome in ocular sarcoidosis: retrospective evaluation of risk factors. Eur J Ophthalmol. 2011;21(6):802-10. 3. Birnbaum AD, Oh FS, Chakrabarti A, et al. Clinical features and diagnostic evaluation of biopsy proven sarcoidosis. Arch Ophthalmol. 2011;129(4):409-13. 4. Jap A, Chee SP. Viral anterior uveitis. Curr Opin Ophthalmol. 2011;22(6):483-8. 5. Van Gelder RN, Prasad AG. Review of Uveitis. Thorofare, NJ; Slack Inc; 2008. 6. Mackensen F, Smith J, Rosenbaum J. Enhanced recognition, treatment and prognosis of tubulointerstitial nephritis and uveitis syndrome. Ophthalmology. 2007;114(5):995-9. 7. Brewerton DA, Hart FD, Nicholls A, et al. Ankylosing spondylitis and HLA-27. Lancet. 1973;1:904-7. 8. Ramsay A, Lightman S. Hypopyon uveitis. Surv Ophthalmol. 2001;46(1):1-18. 9. Khan MA. Race related differences in HLA association with ankylosing spondylitis and Reiters disease in American blacks and whites. J Nat Med Assoc. 1978;70(1):41-2. 10. Smet MD, Ongkosuuwito JV. HLA-B27 related uveitis. In: Krachmer JH, Mannis MJ, Holland EJ, eds. Cornea. 2nd ed. St. Louis: Mosby; 2004:1375-81. 11. Kotaniemi K, Savolainen A, Karma A, Aho K. Recent advances in uveitis of juvenile idiopathic arthritis. Sur Ophthalmol. 2003;48:489-502. 12. Tugal-Tutkun I, Quartier P, Bodaghi B. Disease of the year: juvenile idiopathic arthritis-associated uveitits–classificaiton and diagnostic approach. Oc Immunol Inflamm. 2014;24(1):56-63. 13. Hu-Torres S, Foster CS. Disease of the year: juvenile idiopathic arthritis – differential diagnosis. Oc Immunol Inflamm. 2014;22(1):42-55. 14. Chatzistefanou K, Markomichelakis NN, Christen W, et al. Characteristics of uveitis presenting for the first time in the elderly. Ophthalmology. 1998;105(2):347-52. 15. Zamiri S, Boyd S, Lightman S. Uveitis in the elderly – is it easy to identify the masquerade? Br J Ophthalmol. 1997;81(10):827-31. 16. Santos C. Herpes simplex uveitis. Bol Asoc Med P R. 2004;96(2):71-4. 17. Kawagoe T, Mizuki Nobuhisa. Sarcoidosis. Curr Opin Ophthalmol. 2011;22(6):502-7. 18. Herbort CP, Rao NA, Mochizuki M, et al. International criteria for the diagnosis of ocular sarcoidosis: results of the first international workshop on ocular sarcoidosis. Oc Immunol Inflamm. 2009;17:160-9. 19. Birnbaum A, Little DM, Tessler HH, Goldstein DA. Etiologies of chronic anterior uveitis at a tertiary referral center over 35 years. Oc Immunol Inflamm. 2011;19:19-25. 20. Rosenbaum JT. Round 20: Why a Rheumatologist needs to Understand Uveitis. Johns Hopkins Arthritis Center. Available at www.hopkinsarthritis.org/physician-corner/rheumatology-rounds/round-20-why-a-rheumatologist-needs-to-understand-uveitis/. Accessed August 18, 2016. 21. Evans M, Sharma O, LaBree L, et al. Differences in clinical findings between caucasians and african americans with biopsy proven sarcoidosis. Ophthalmology. 2007;114(2):325-33. 22. Lobo A, Barton K, Minassian D, et al. Visual loss in sarcoid related uveitis. Clin Exp Ophthalmol. 2003;31(4):310-6. 23. Barile G, Flynn T. Syphilis exposure in patients with uveitis. Ophthalmology. 1997;104(10):1605-9. 24. Gaudio PA. Update on ocular syphilis. Curr Opin Ophthalmol. 2006;17(6):562-6. 25. Chao JR, Khurana RN, Fawzi AA, et al. Syphilis: reemergence of an old adversary. Ophthalmology. 2006;113(11):2074-9. 26. Yap SC, Tan YL, Chio MT, Teoh SC. Syphilitic uveitis in a singaporean population. Oc Immunol Inflamm. 2014;22(1):9-14. 27. Emmons WW, Church LW. Syphilitic uveitis. West J Med. 1994;161(2):168-71. 28. Cunningham E, Eandi CM, Pichi F. Syphilitic uveitis. Oc Immunol Inflamm. 2014;22(1):2-3. 29. Amaratunge BC, Camuglia JE, Hall AJ. Syphilitic uveitis: a review of clinical manifestations and treatment outcomes in human immunodeficiency virus-positive and negative patients. Clin Exp Ophthalmol. 2010;38(1):68-74. 30. Gupta A, Bansal R, Gupta V, et al. Ocular signs predictive of tubercular uveitis. Am J Ophthalmol. 2010 Apr;149(4):562-70. 31. Shakarchi FI. Ocular tuberculosis: current perspectives. Clin Ophthalmol. 2015;9:2223-7. 32. Tabbara KF. Tuberculosis. Curr Opin Ophthalmol. 2007;18(6):493-501. 33. Mohamed Q, Zamir E. Update on Fuchs’ uveitis syndrome. Curr Opin Ophthalmol. 2005;16(6):356-63. 34. Velilla S, Dios E, Herreras JM, Calonge M. Fuchs’ heterochromic iridocyclitis: A review of 26 cases. Oc Immol Inflamm. 2001;9(3):169-75. 35. La Hey E, de Jong PTVM, Kijlstra A. Fuchs; heterochromic cyclitis:review of the literature on pathogenic mechanisms. Br J Ophthalmol. 1994;78:307-12. 36. de Groot-Mijnes JD, de Visser L, Rothova A, et al. Rubella virus is associated with fuchs heterochromic iridocyclitis. Am J Ophthalmol. 2006;141(1):212-4. 37. Birnbaum AD, Tessler HH, Schultz KL, et al. Epidemiologic relationship between fuchs heterchromic iridocyclitis and the United States rubella vaccination program. Am J Ophthalmol. 2007;144(3):424-8. 38. Tabbot BR, Tessler HH, Williams D. Fuchs’ heterochromic iridocyclitis in Blacks. Arch Ophthalmol. 1988;106:1688-90. 39. Rothva A, Ooijman F, Kerkhoff F, et al. Uveitis masquerade syndromes. Ophthalmology. 2001;108(2):386-99. 40. Sharma T, Grewal J, Gupta S, Murray PI. Ophthalmic manifestations of acute leukaemias: the ophthalmologist’s role. Eye (Lond). 2004;18:663-72. 41. Byles DB, Frith P, Salmon JF. Anterior uveitis as a side effect of topical brimondine. Am J Ophthalmol. 2000;130:287-91. 42. Wakefield, Chang JH, Amjadi S, et al. What is new HLA-B27 acute anterior uveitis? Ocul Immunol Inflamm. 2011 Apr;19(2):139-44. 43. Rosenbaum JT, Wernick R. The utilitiy of routine screening of patients with uveitis for systemic lupus erythematosus or tuberculosis. Arch Ophthalmol. 1990;108:1291-3. 44. Gallager K, Viswanathan A, Okhravi N. Association of systemic lupus erythematosus with uveitis. JAMA Ophthalmol. 2015;133:1190-3. |