Corneal Harbingers of Systemic Disease
A routine slit lamp exam may reveal telltale findings of health concerns elsewhere in the body. Here are the most common to look for.
Goal Statement:
Although ocular manifestations of systemic diseases most often present with retinal involvement, several systemic conditions—such as diabetes, autoimmune disorders and infectious diseases—can be diagnosed via corneal presentation. This article educates optometrists on how to recognize the corneal signs of systemic diseases and understand their role in disease management.
Faculty/Editorial Board:
Aaron Bronner, OD
Credit Statement:
This course is COPE approved for 2 hours of CE credit. COPE ID 45159-SD. Please check your state licensing board to see if this approval counts toward your CE requirement for relicensure.
Joint-Sponsorship Statement:
This continuing education course is joint-sponsored by the Pennsylvania College of Optometry.
Disclosure Statement:
Dr. Bronner has no relevant financial relationships to disclose.
Though retinal vasculature disorders or iritis come to mind first when we think about systemic disease and the eye, the cornea—with its unique clarity and dual zones of immune function—provides a unique window into a wide variety of systemic diseases that otherwise would require laboratory or imaging-based evaluation. Systemic manifestations in the cornea range from the mundane, as with arcus senilis, to the unusual and life-threatening, as with multiple myeloma or vasculopathic disease. Reviewing many of these manifestations—ignoring the well-described or general associations, such as hypercholesterolemia with arcus and keratitis sicca associ ated with its myriad autoimmune diseases—will ensure you are prepared to see even the most unusual corneal issues stemming from systemic disease.
Diabetes Mellitus
One of the most important systemic diseases monitored by optometrists for its ocular effects is diabetes. We are all aware of the retinopathy and neovascularization of the iris and/or the angle with potential for subsequent glaucoma, as well as the severe extra-ocular effects of diabetes; namely, nephropathy and neuropathy. Given that the eye is such a prominent site for diabetic involvement and in light of the potential for the disease to manifest as neuropathy, it should come as no surprise that the cornea, which is the most densely innervated tissue in the body, may also be impacted by diabetes.
Diabetic keratopathy is thought to manifest as a result of changes in the density of corneal innervation, particularly within the sub-basal nerve plexus. Based on the results of at least one study group assessing Type I diabetes, these changes result in reduced corneal sensation, increasing corneal thickness and cellular polymorphism of both the corneal epithelium and endo-thelium. What's more, the level of diabetic keratopathy correlates well with the levels of retinopathy, systemic polyneuropathy and nephropathy.1,2
While these changes can be assessed to some degree with formal esthesiometry and pachymetry, diabetic corneal neuropathy can only be objectively defined by confocal microscopy and so falls outside the optometrist's ability to quantify. It is useful to know, however, that as the disease progresses, patients with diabetes may become more neurotrophic and develop thicker corneas. In the study referenced, patients without diabetes had an average corneal thickness of 526μm, patients with mild neuropathy had 558μm and patients with severe neuropathy had corneal thickness of 625μm.1
Corneal Lines Comparison |
||
 Waite-Beetham lines in a 73-year-old diabetic. |
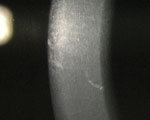 Vogt's striae in a 31-year-old keratoconic patient. Note how much finer, more tightly organized and more vertically arranged the Vogt's striae are compared to Waite- Beetham lines. |
 Corneal stromal edema. Edematous lines are much broader and less organized than Waite-Beetham lines. |
A frequently encountered but infrequently recognized ocular manifestation of diabetes is the presence of pleats in either the deep stroma or Descemet's membrane of the non-ectatic eye—sometimes simply referred to as Descemet's wrinkles and formally known as Waite-Beetham lines. They are more prominent, though arranged with less density and precision, than Vogt's striae seen in keratoectasias. They are also not as thick and irregular as pleats seen in edematous corneas.
Descemet's wrinkles may be single or multiple, are longer than Vogt's striae as seen in keratoconus, and tend to be somewhat wavy, though generally are arranged vertically or, less commonly, obliquely (unlike edematous pleats, which may be arranged in any direction).
These disturbances do not seem to create pathology of the cornea, nor do they appear to be the harbinger of worsening systemic disease. In fact, while Descemet's wrinkles are more common in diabetes (27% to 38% prevalence), they occur in unaffected patients as well (8% to 10.5%).3,4 They become more common with age in both populations, and occur earlier in patients with diabetes.3 Their presence—particularly in a patient for whom you may already suspect diabetes—may be a warning sign that a patient has developed the condition.
Peripheral Ulcerative Keratitis
Isolated corneal effects of iritis, scleritis and episcleritis—often associated with autoimmune disease—are also possible, and given their significant relationship with ocular morbidity and overall mortality, it is important for doctors to be aware of them.
The cornea can be roughly thought of as having two distinct immune zones:
• The central cornea, characterized by its avascularity and lack of lymphatics, is relatively remote from the immune response and therefore sheltered to a certain degree, from destructive systemic infectious as well as autoimmune processes.
• The peripheral cornea, however, with its proximity to the vasculature, lymphatic channels and secondary lymph tissue of the conjunctiva, sclera and episclera, is in a prime location to manifest immune processes associated with systemic infectious and autoimmune disease. Peripheral ulcerative keratitis (PUK) is a significant ocular manifestation of systemic vasculitides, connective tissue diseases and infectious disease of the juxtalimbal cornea.
Systemic Etiologies of PUK
|
While there are a number of other corneal manifestations of autoimmune disease more common than PUK, it remains a very important pathology to be aware of because of its potential for being the presenting manifestation of autoimmune disease; one study showed it preceded systemic diagnosis in 28% of cases.5-8 Additionally, among some forms of the disease it is associated with a mortality rate as high as 50% over five years if systemic immunomodulatory agents are not implemented.5-8
Though many autoimmune or systemic infectious diseases may result in PUK, the underlying mechanism of a perilimbal vasculitis seems to be relatively uniform. Characterized by a crescentic infiltrate with an overlying epithelial defect along the peripheral cornea, PUK lesions will progressively thin in the absence of appropriate treatment and may lead to perforation of the cornea.
Though it's most typically associated with adjacent areas of scleritis, PUK may occur as a primary manifestation as well; when isolated to the cornea without any systemic etiology it is known as a Mooren's ulcer, though some argue that Mooren's ulcer is actually associated with hepatitis.5,9
As topical corticosteroids are contraindicated due to their possible promotion of collagenase activity (thereby enhancing keratolysis), topical treatment is limited primarily to aggressive lubrication. Surgical and parasurical procedures such as application of a bandage contact lens, tissue glue, conjunctival resection and keratoplasty may temporize any risk of perforation or even act to stunt the ulcerative process, but ultimately systemic treatment is needed to abate the attack and reduce risk of recurrence.6 Though matrix metalloproteinases are implicated in the disease process, adjunctive use of tetracyclines is often ineffective.
In the treatment of autoimmune-associated PUK, it is imperative to recognize the condition's role as a herald of worsening systemic disease; widespread vasculopathic dis ease can lead to the patient's death. In addition to difficult treatment of their ocular disease, which may generally be managed with oral cortocosteroids, these patients need to be placed on potent cytotoxic or immunomodulatory agents to control their underlying condition.7
Corneal Deposits
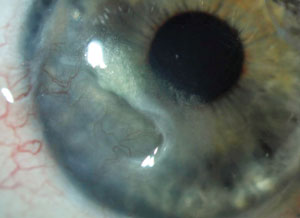 Active peripheral ulcerative keratitis in a 35-year-old with thinning, vascularization and lipid deposit. |
Perhaps no corneal manifestation of systemic disease is as recognizable as the verticillata seen with the use of the anti-arrhythmic medication amiodarone. These gray to golden whorled deposits in the interpalpebral region of cornea occur almost universally in patients on this drug and generally dissipate upon cessation of the medication.10 The rate of appearance upon initiation of the medication (days to months) and the disappearance upon cessation (months to years) varies widely from individual to individual.10
While quite prominent with biomicroscopy, verticillata generally have no effect on vision beyond occasional reports of glare and halos. Amiodarone is the most common source of verticillata, but there are other sources as well, including chloroquinine, hydroxychloroquine—where, in the setting of corneal deposits, there is also a higher rate of retinopathy—tamoxifen, phenothiazine, suramin and indomethacin, among others.10,11
While it may seem confusing for this widely varied group of medications to share the potential for creating verticillata, it is not their respective mechanisms that result in the manifestation, but the biochemical behavior of the molecules. They all share ampiphilic properties, which allows the molecules to penetrate and create lipid-based complexes within lysosomes.10 This eventually leads to the whorl keratopathy.
These lysosome-born deposits are generally held in the basal epithelium. Confocal microscopy has also identified abnormalities within the anterior stroma. The deposits and other abnormalities appear to develop as medication precipitates through the tear film, a reason why concomitant contact lens use may result in more dramatic whorls.10,12
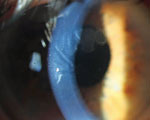
 Corneal crystals in a 56-year-old man with multiple myeloma. While they appear granular in this direct illumination photo, the crystals scintillated with movement of light across their surface. |
Beyond pharmaceuticals, Fabry's disease—an X-linked recessive disorder of lipid metabolism and one of many metabolic diseases that results in corneal deposits—has a near-universal link to verticillata as well.13 Though the verticillata seen with Fabry's tend to display more well-formed vortices (whereas drug-induced variants have more of a "cat's whisker appearance"), the underlying deposit, again, is lipid complexes within lysosomes of the deep epithelium.10,13
While the link between Fabry's and corneal deposits is very strong, the chances of diagnosing the condition based on ocular exam is quite low due to the limited symptomology associated with its ocular manifestations. In fact, in a study that looked at the utility of community ocular screening for Fabry's disease, only one case was discovered in over eight million patients—a fact that led the authors to question the utility of community eye screenings for this disease. The authors, however, then go on to point out how important it is to seize these rare opportunities to recognize the findings of verticillata when they could potentially yield a diagnosis of Fabry's.14
Though much less common than verticillate corneal deposits, corneal crystals are important to be aware of given their association with potentially fatal systemic conditions. Beyond localized crystalline corneal dystrophies such as Schny-der's and Bietti's corneo-retinal dystrophy, metabolic disease, neoplastic and pre-neoplastic disorders are other sources of corneal crystals.
Cystinosis is an X-linked recessive metabolic disease of the amino acid cystine, which becomes trapped in lysosomes, potentially affecting tissue function systemically. There are three types of cystinosis, classified based on the time of development: infantile, intermediate and adult-onset, with infantile being the most severe and possible fatal. The corneal crystals that develop regardless of subtype are generally fine and, given sufficient time, are diffusely distributed throughout the anterior cornea. Though they may be very prominent, a patient's visual acuity is generally not affected, barring photophobia.15
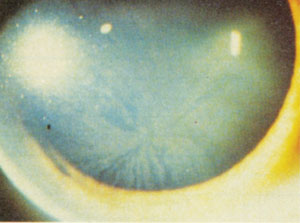 Fabry's associated corneal verticillata. Note how much more well formed the pattern is than with typical medication-associated vortices. Photo: Genzyme Corp |
Corneal crystals that develop in middle age may be caused by either local dystrophy or systemic metabolic disease. This group of patients must also have plasma cell dyscrasias on the differential diagnosis. Plasma cell dyscrasias—which include multiple myeloma, monoclonal gammopathy of undetermined significance (MGUS) and Waldenstrom's macroglobunemia, among others—are characterized by an atypical population of plasma or B cells, which redundantly produce a single, monoclonal form of immunoglobulin (IG) or an IG fragment, which then may become deposited in tissue.
The distribution and exact appearance of the corneal crystals seen in patients with these conditions vary widely: from diffuse fine through-and-through crystals, to very localized needle-like changes, to lattice and granular-like deposits and even those with verticillate appearance.11,16-18 They generally share the common feature of being crystalline and iridescent upon close examination.16 Despite the clear cornea being an ideal location to see systemically-derived protein deposits, crystals are not common findings, with one study reporting only a 1% incidence in patients with MGUS.16
Though an uncommon manifestation of uncommon systemic diseases, corneal crystals in this setting have diagnostic value, as symptoms of these diseases are relatively non-specific, and the presence of corneal crystals can help steer the clinician toward an important diagnosis. In the absence of other diagnostic clues, it may justify a hematology referral.
Band Keratopathy
Metabolic Dysfunction and the CorneaThere are close to 40 recognized lysosomal storage diseases that may result in ocular findings, many of which will generate cornea-specific findings. All of these disorders share the common mechanism of metabolic products becoming trapped in lysosomes due to lack of a variety of metabolic enzymes. For more on metabolic diseases affecting the cornea, Holland's Cornea (specifically, chapter 64 by Kenyon, Navon and Haritoglou) covers this broad group of disorders and their wide range of possible effects on the cornea.15 |
A frequently encountered sequela of chronic ocular inflammation, band keratopathy is characterized by a sub-Bowman's layer opacification via deposition of calcium phosphate. In addition to being caused by various ocular pathologies, systemic diseases that cause hypercalcemia may also lead to band keratopathy.19
In hyperparathyroidism, which results in systemic hypercalcemia, band keratopathy may be among the presenting signs. Interestingly, as opposed to ocular band keratopathy, this keratopathy may spontaneously regress when the underlying etiology has been treated. Other systemic etiologies of band keratopathy are sarcoidosis, gout, chronic renal failure, multiple myeloma and metastatic disease.19
In general, band keratopathy in an otherwise unremarkable eye should generate an appropriate level of clinical suspicion that systemic hypercalcemia may be playing a role. These patients should be asked about symptoms of bone pain or frequent urination, and be sent for urinalysis and blood tests.
Infection
Although corneal manifestations of endogenous infectious disease are rare, given the significance of their underlying etiologies it's important for clinicians to be aware of them. Perhaps the most well-described form is phlyctenular keratoconjunctivitis and its association with systemic Mycobacterium tuberculosis infection.
Local and Systemic Causes of Corneal Phlyctenules
|
Corneal phlyctenules are nodular lesions made up of lymphoid tissue; they may either be limbal or, when on the clear cornea, remain attached by a vascular tuft to the limbus. Their nodules may be smooth or ulcerated depending on their staging, and when isolated to the limbus they leave no clear zone between limbus and cornea.
The lesions represent a delayed-onset hypersensitivity reaction to a wide variety of microbial and viral antigens, the most common being blepharitis associated with Staphylococcus.20 When associated with tuberculosis, these will occur almost exclusivity at a young age, as the body eventually becomes desensitized to the causative antigen as it ages.20
Though the precise etiology will dictate treatment, all phlyctenules will respond locally to topical corticosteroids. As phlyctenules are a hypersensitivity reaction, the topical use of corticosteroids is safe and in no way contraindicated by the underlying infectious etiology, though of course when a systemic etiology is suspected concurrent systemic treatment is necessary.20,21
A second mycobacterial infection, Mycobacterium leprae, is actually reported to have the highest rate of ocular involvement in an infected individual of any systemic infectious disease, with estimates of ocular involvement being as high as 100% in an untreated population.22,23 Though the worldwide burden has been significantly reduced with effective multidrug therapy (MDT) and the disease is currently most associated with developing countries, leprosy does still constitute a significant worldwide source of ocular morbidity. In the United States, a country of immigrants, it is something with which the eye care provider should at least have passing familiarity.
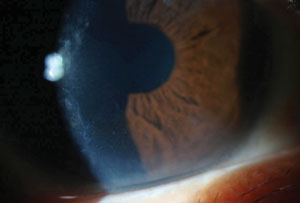
 Leprosy keratitis in a patient with previously undiagnosed leprosy who presented to clinic in the US for cataract evaluation. |
As the organism has a predilection for cooler temperatures, the anterior segment and adnexa, which are 4° to 6° C cooler than the deep orbit, are the affected tissues. Nearly all other forms of ocular disease associated with systemic infection generally show predilection for the posterior segment; however, whether leprosy affects the posterior segment at all is a matter of debate.24 The sight-threatening impact of leprosy generally has to do with lagophthalmos resulting in corneal opacification or chronic plastic iritis resulting in gradual destruction of the ciliary body; however, direct involvement of the cornea is not uncommon.
While most corneal findings in leprosy are general—punctate epithelial erosions, prominence of corneal nerves (with greater specificity if beading of nerves occurs) and varying degrees of neurotrophy— the subepithelial infiltrate seen with leprosy is nearly pathognomonic, sharing a similar appearance with the perineuritis seen in Acanthamoeba keratitis.23 They manifest as focal chalky-white, pinhead-sized subepithelial opacities within an area of avascular pannus, and unlike similar lesions seen with Acanthamoeba, which usually begin centrally, those seen with leprosy most often begin in the superior cornea.25,26 These lesions represent accumulations of active bacillus within "foam cell" monocytes; however, given the down-regulation of the immune response to M. leprae over time, they display little to no local infiltration of white cells.27 Further, as no cuture media exists for M. leprae, they cannot be effectively processed for microbiologic studies.
Treatment regimens for ocular disease resulting from leprosy is similar to that of tuberculosis: local corticosteroids are fine when indicated, but refer to infectious disease for confirmation of diagnosis (in this case with skin biopsy) and initiation of multidrug therapy.
Syphillis
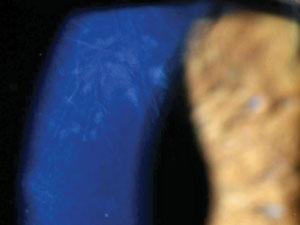 Deep interstitial ghost vessels in a patient with contact lens induced keratitis mimicking syphilitic keratitis. |
Interstitial keratitis associated with syphilis is among the most widely recognized corneal effects of infectious systemic disease. Syphilitic keratitis, in and of itself, is an atypical manifestation of an increasingly uncommon disease, accounting for only 5% of all cases of syphilitic eye disease, with most of these cases being caused by the congenital form of the disease.15 Though the rate of acquired syphilis is increasing again, the congenital disease remains uncommon, affecting only one in one million.15,28 Bilateral presentation is most frequently associated with the congenital disease, as is the tendency to develop ghost vessels upon resolution.
Though the term "congenital syphilis" may lead the practitioner to view this disease as one of gestation and infancy, syphilitic interstitial keratitis is a late manifestation of congenital disease that commonly develops between the ages of five and 20. Likewise, with the acquired disease, keratitis is most typically a late manifestation, occurring years after the original infection.
In both congenital and acquired disease, the keratitis initially creates a deep focal inflammation of the stroma, typically starting in the periphery with a slight predilection for the superior cornea. In congenital cases, this will sequentially develop within months in the fellow eye. Vascularization typically follows the initial inflammation and perpetuates it, before ultimately burning out and regressing.
In acquired syphilis, the initial keratitis resembles congenital disease, though tends to remain less severe, unilateral and avascular.15
Though uncommon—and in the acquired form limited primarily to those who practice risky sexual behaviors or intravenous drug use—syphilis should be suspected in any case of bilateral stromal keratitis or deep stromal keratitis without a previous episode of more superficial disease (i.e., if it's not herpetic).
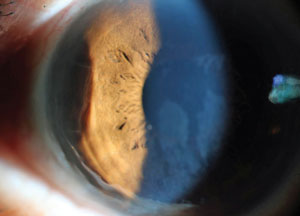 Peripheral thinning in a patient previously treated for syphilitic peripheral ulcerative keratitis. Photo: Jeff Urness, OD. |
Unfortunately, patients without a previous diagnosis do not tell you they have congenital syphilis and I've yet to have a patient write "risky sexual behavior" or voluntarily document "intravenous drug user" on their initial health intake form. Because of this lack of transparency, serologic testing for syphilis should be ordered in cases where clinical suspicion puts syphilis on a differential.15,28 But keep in mind that there are a number of non-syphilitic causes of interstitial keratitis and corneal ghost vessels, with some being as simple as contact lens use. Within the United States, herpetic disease is twice as likely as any other pathology to cause the presentation.29
While this review is far from complete—with important manifestations such as copper deposits in the corneal periphery as seen with Wilson's disease being omitted—hopefully it serves as a useful review and reminder of the spectrum and possible seriousness of these manifestations both locally to the cornea itself and systemically. It's important to periodically be reminded that although our focus and attention is on the organ of the eye, the overall health of the patient is ultimately the most important consideration for us as optometrists. Where ocular and systemic diseases converge, it is our responsibility as the nation's primary eye care provider to be astute in our observation and conscientious in our assessments so we can refer patients in an appropriate and timely manner. You might just be saving someone's life!
Dr. Bronner is a staff optometrist at the Pacific Cataract and Laser Institute of Kennewick, Wash.
References
- Rosenberg M, et al. Corneal structure and sensitivity in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci. 2000;41:2915-21.
- Petropoulos IN, et al. Rapid automated diagnosis of peripheral neuropathy with in vivo corneal confocal microscopy. Invest Ophthal-mol Vis Sci. 2014;55:2071-8.
- Henkind P, Wise GN. Descemet's wrinkles in diabetes. Am J Ophthalmol. 1961;52:371-4.
- 4. Mocan MC, Irkc M, Orhan M. Evidence of Waite-Beetham lines in the corneas of diabetic patients as detected by in vivo confocal microscopy. Eye. 2006;20:1488-90.
- Ladas JG, Mondion BJ. Systemic disorders associated with peripheral corneal ulceration. Current Opinion in Ophthalmology. 2000;11:468-71.
- Messmer EM, Foster CS. Vasculitic Peripheral Ulcerative Keratitis. Survey of Ophthalmology. 1999;43:379-96.
- Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripherial ulcerative keratitis: Effects of systemic immune suppression. Ophthalmology. 1984;91:1253-63.
- Tauber J, et al. An Analysis of therapeutic decision making regarding immunosuppressive chemotherapy for peripheral ulcerative keratitis. Cornea.1990;9:66-73.
- Wilson SE, et al. Mooren's ulcers and hepatitis C virus Infection. N Engl J Med. 1993;329:62.
- Hollander DA, Aldave AJ. Drug-induced corneal complications. Current Opinion in Ophthalmology. 2004;15:541-8.
- Sharma P, et al. Cloudy corneas as an initial presentation of multiple myeloma. Clinical Ophthalmology. 214;8:813-7.
- Falke K, et al. The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratophaty: a confocal laser scanning microscopy study. Grafes Arch Clin Exp Opthalmol. 2009;247:523-34.
- Sodi A, Ioannidis A, Pitz S. Ophthalmological manifestations of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11599/.
- Hauser AC, et al. Results of an ophthalmologic screening programme for identification of cases with Anderson-Fabry disease. Ophthalmiogica. 2004;218:207-9.
- Kenyon KR, Navon SE, Haritoglou C. Corneal manifestations of metabolic disease. In: Krachmer JH, Mannis MJ, Holland EJ, eds. Cornea. 2nd ed. St. Louis: Mosby;2004:749-76.
- Spiegel P, et al. Unusual presentation of paraproteinemic corneal Infiltrates. Cornea. 1990;9:81-5.
- Lisch W, et al. Chameleon-like appearance of immunotactoid keratopathy. Cornea. 2012;31:55-58.
- Chou JL, Sink ML. Corneal crystals: a precursor to cancer. Optometry and Vision Science. 2011;88:E543-7.
- Mora ML, Smith RE. Corneal and systemic diseases. In: Tasman W, Jaeger EA eds. Duane's Ophthalmology. Lipincott Williams Wilkins; 2006.
- Mondino BJ. Inflammatory diseases of the peripheral cornea. Ophthalmology. 19988;95:463-72.
- Rapuano C, Luchs JI, Kim T. Corneal infections, inflammations, and surface disorders. In: Anterior Segment; The Requisites in Ophthalmology. Mosby; 2000:115-8.
- Dethlefs R. Prevalence of ocular manifestations of leprosy in Port Moresby, Papua New Guinea. British Journal of Ophthalmology. 1981;65:223-5.
- Choyce DP. Ocular leprosy, with reference to certain cases shown. Proceedings of the Royal Society of Medicine. 1955;48(2):108-12.
- Ffytche TJ. Role of iris changes as a cause of blindness in lepro matous leprosy. British Journal of Ophthalmology. 1981;65:231-9.
- Choyce DP. Diagnosis and management of ocular leprosy. British Journal Ophthalmology. 1969;53:217-23.
- Krachmer JH, Palay D. Corneal Atlas. 2nd 466 Edition. Mosby Elsevier; 2006.
- Myrvang B, et al. Immune responsiveness to mycobacterium leprae and other mycobacterial antigens throughout the clinical and histopathological spectrum of leprosy. Clinical Expir. Immunology. 1973;14:541-53.
- Wilhemus KR. Syphilitic interstitial keratitis. In: Krachmer JH, Mannis MJ, Holland EJ, eds. Cornea. 2nd ed. St. Louis: Mosby; 2004:1133-59.
- Schwartz GS, Harrison AR, Holland EJ. Etiology of immune stro-mal (interstitial) keratitis. Cornea. 1998;17(3):278-81.