 |
Are You Up to Speed on Inherited Retinal Dystrophies?
Enhance your patient care with an improved understanding of these genetic disorders and how they present in clinical practice.
By Roya Attar, OD, Rachel Steele, OD, and Jim Williamson, OD
Jointly provided by the Postgraduate Institute for Medicine (PIM) and the Review Education Group
Release Date: June 15, 2024
Expiration Date: June 15, 2027
Estimated Time to Complete Activity: two hours
Target Audience: This activity is intended for optometrists who want to learn about the impact this system has on practice.
Educational Objectives: After completing this activity, participants should be better able to:
Recognize the presentations of various inherited retinal dystrophies.
Effectively diagnosis inherited retinal dystrophies in clinical practice.
Recognize the value and limitations of genetic testing for these patients.
Determine which IRD patients could benefit from genetic testing.
Disclosure of Conflicts of Interest: PIM requires faculty, planners and others in control of educational content to disclose all their financial relationships with ineligible companies. All identified conflicts of interest are thoroughly vetted and mitigated according to PIM policy. PIM is committed to providing its learners with high-quality, accredited CE activities and related materials that promote improvements or quality in health care and not a specific proprietary business interest of an ineligible company.
Those involved reported the following relevant financial relationships with ineligible entities related to the educational content of this CE activity: Faculty - Drs. Steele and Williamson have nothing to disclose. Dr. Attar is on advisory boards for Heidelberg Engineering, Apellis and OcuTerra Therapeutics. Planners and Editorial Staff - PIM has nothing to disclose. The Review Education Group has nothing to disclose.
Accreditation Statement: In support of improving patient care, this activity has been planned and implemented by PIM and the Review Education Group. PIM is jointly accredited by the Accreditation Council for Continuing Medical Education, the Accreditation Council for Pharmacy Education and the American Nurses Credentialing Center to provide CE for the healthcare team. PIM is accredited by COPE to provide CE to optometrists.
Credit Statement: This course is COPE-approved for two hours of CE credit. Activity #128547 and course ID 91657-TD. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure of Unlabeled Use: This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. The planners of this activity do not recommend the use of any agent outside of the labeled indications. The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of the planners. Refer to the official prescribing information for each product for discussion of approved indications, contraindications and warnings.
Disclaimer: Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient’s condition(s) and possible contraindications and/or dangers in use, review of any applicable manufacturer’s product information and comparison with recommendations of other authorities.
Inherited retinal dystrophies (IRDs) encompass a group of genetic disorders affecting the retina, often leading to progressive vision loss. Their prevalence hovers around one in 1,400 individuals, which means most practitioners will see at least several per year. Some IRDs masquerade as other disorders, while others are clinically obvious. Genetic testing doesn’t always offer confirmation, but when it does, it provides insight into the prognosis, inheritance pattern, current or future trials and possible syndromic features. Optometrists should recognize these entities and their nuanced characteristics to guide their testing strategy and arrive at the correct diagnosis.1
Stargardt’s Disease (STGD1)
This condition is the most commonly inherited juvenile retinal dystrophy, with an estimated incidence of one in 8,000 or one in 10,000.2 The inheritance pattern is autosomal recessive, requiring biallelic variations in the ABCA4 gene, which has also been implicated in childhood-onset cone-rod dystrophy, bull’s eye maculopathy and RP.2
The ABCA4 gene codes for an ATP-binding cassette transporter that plays a role in the visual cycle in recycling all-trans-retinal.3 When dysfunctional, this protein leads to accumulation of N-retinylidene-N-retinyl-ethanolamine (A2E) within the retinal pigment epithelium (RPE), resulting in eventual photoreceptor cell death.3,4 Currently, there have been over 2,200 ABCA4 variations reported.4
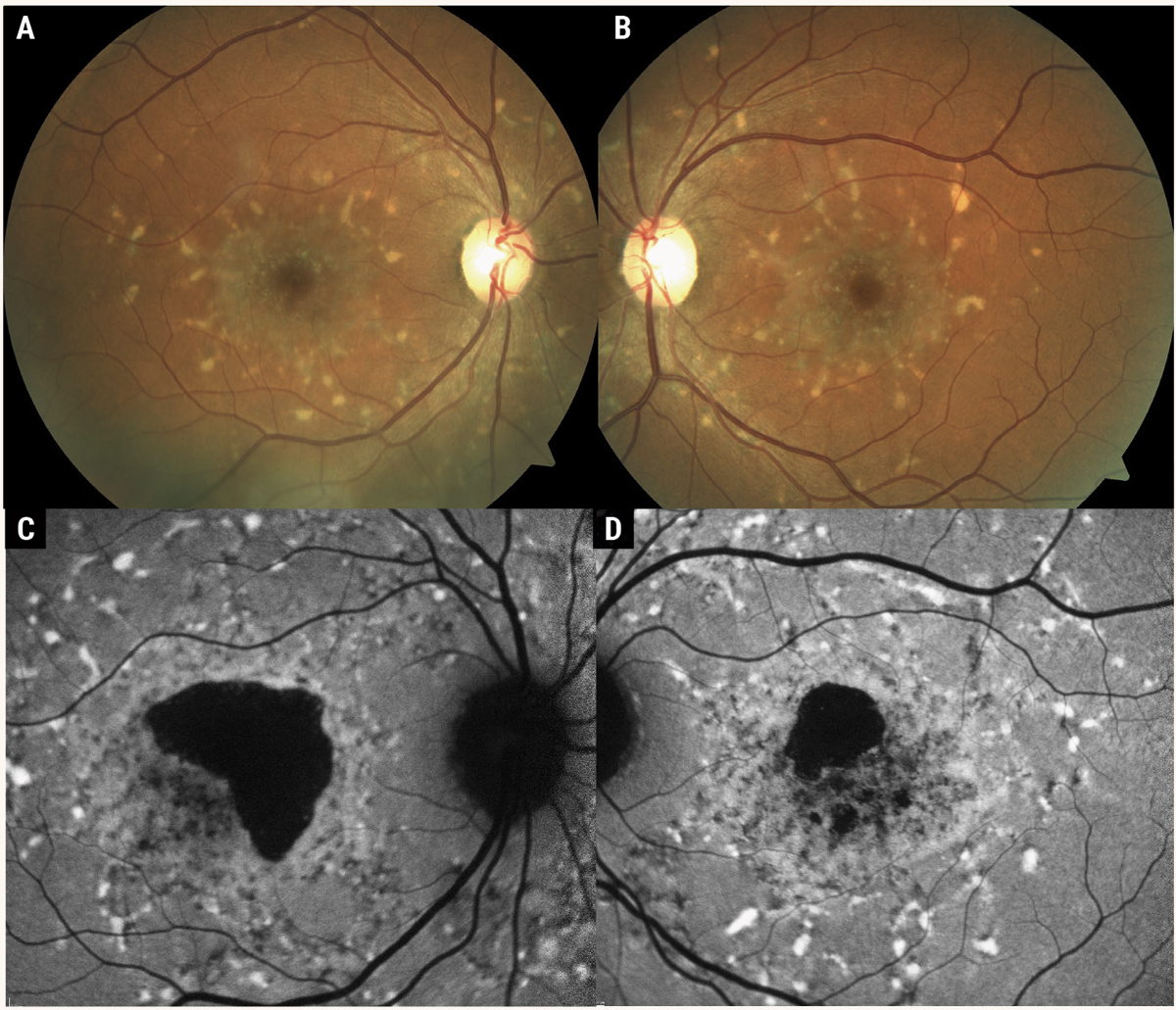
While ABCA4 disease can manifest in a range of phenotypes, STDG1 classically produces a beaten-bronze fundus appearance with whitish, yellow subretinal pisciform flecks scattered throughout the posterior pole with varying degrees of photoreceptor atrophy (Figure 1). The age of onset may vary from early-onset to early-adult onset to late-onset; however, the average age is about 15 years.5
 |
|
Fig. 1. Color fundus photographs (A, B) of Stargardt’s disease. These photos demonstrate the classic STDG1 phenotype of white, pisciform flecks scattered throughout the posterior pole with the presence of atrophic macular lesions. Fundus autofluorescence (C, D) demonstrates the hyper-autofluorescent nature of the pisciform flecks, in contrast to drusen, which are often iso-autofluorescent. Photos: Mohammad Rafieetary, OD. Click image to enlarge. |
Early-onset ABCA4 disease is associated with inheritance of two severe variants of the gene and may progress to severe vision loss early in life due to extensive outer retinal atrophy.5 Late-onset disease is thought to be caused by one severe variant and one mild variant of the ABCA4 gene and generally results in less severe disease. However, it can result in extrafoveal outer retinal atrophy, which may be misdiagnosed as geographic atrophy from AMD.4
In up to a quarter of cases, patients with early-onset disease may have reduced vision with no abnormalities detectable on clinical examination.6 Multimodal imaging can reveal early, subtle findings, including thickening of the foveal or parafoveal external limiting membrane on OCT. Fundus autofluorescence (FAF) may show hyper-autofluorescent flecks or general hyper-autofluorescence that might not be apparent on clinical examination.
OCT findings of more advanced disease include thinning of the ONL, loss of the ellipsoid zone (EZ) and RPE atrophy. Pisciform flecks appear on OCT as hyperreflective subretinal deposits and hyper-reflective foci, which may disrupt the EZ and extend into the inner retinal layers. Early-phase intravenous fluorescein angiography (IVFA) classically reveals a “silent choroid,” where the expected choroidal flush is masked by abnormal accumulation of lipofuscin within RPE cells.
Differentiating STDG1 from masqueraders, such as AMD, is increasingly important with emerging invasive therapies for geographic atrophy (i.e., pegcetacoplan and avacincaptad pegol).4 A distinguishing factor includes the presence of drusen. Drusen are clinically well-defined, round, yellow lesions visible on clinical examination, while the flecks in Stargardt’s are often whitish yellow, with an ill-defined pisciform shape, and may be difficult to discern clinically.
Additionally, drusen are located below the RPE (with the exception of subretinal drusenoid deposits), while flecks are located above the RPE and may disrupt the EZ. Lastly, on FAF drusen are most commonly iso-autofluorescent due to their location below the RPE, while the flecks in STGD1 are strongly hyper-autofluorescent and located above the RPE.
Genetic testing is critical for these patients from the standpoints of patient counseling, predicting disease severity, genetic counseling and identifying eligibility for future gene therapies or clinical trials.
 |
|
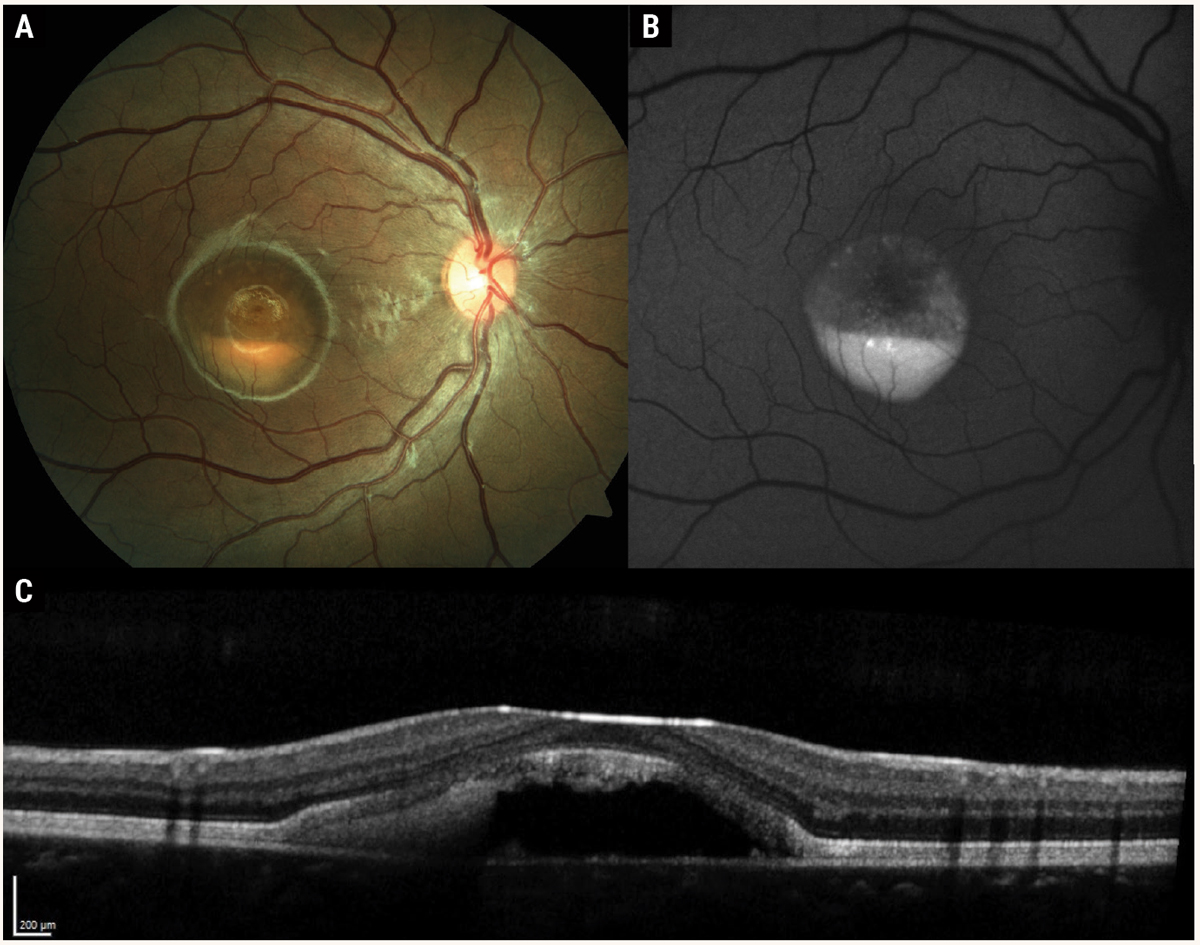
Fig. 2. Multimodal imaging of Best’s disease with subfoveal vitelliform lesion in the “pseudohypopyon” stage. Color fundus photography (A) shows yellow subretinal material gravitating inferiorly, resembling a hypopyon. Fundus autofluorescence (B) reveals hyper-autofluorescence of the vitelliform material. OCT (C ) demonstrates hyperreflective subretinal material with optically empty space superiorly. Note the overlying photoreceptor outer segments are elongated, similar in appearance to chronic central serous chorioretinopathy. Photos: Mohammad Rafieetary, OD. Click image to enlarge. |
Best’s Disease
This autosomal dominantly inherited macular disorder is caused by a mutation in the BEST1 gene (chromosome 11). BEST1 codes for the bestrophin protein, a calcium-gated chloride co-transporter located at the basolateral membrane of RPE cells.7 Currently, there have been over 100 disease causing variants reported in the BEST1 gene. The condition has an estimated prevalence of one in 5,500 in the US, with a bimodal distribution of onset first before puberty and then after puberty.8,9
Five stages of the disease have been defined.7,8,10 In stage 1, the pre-vitelliform phase, the macula has an unremarkable appearance with a reduced Arden ratio (<1.55) on an electrooculogram (EOG). Stage 2 is termed the vitelliform stage, in which there is a one- to two-disc diameter accumulation of yellow subretinal material, resembling an egg yolk.
Stage 3 is the pseudohypopyon stage, where the subretinal material gravitates inferiorly, creating the appearance of a hypopyon in the macula (Figure 2). Stage 4 is the vitelliruptive stage, with variable reabsorption of the vitelliform material creating a “scrambled egg” appearance. Stage 5 represents advanced disease with macular atrophy, subretinal fibrosis or choroidal neovascularization. The prevalence of macular neovascularization (MNV) is estimated to be about 5.7%.10
On clinical examination, fundus findings vary depending on the disease stage. Multimodal imaging is useful in describing the clinical course of Best’s disease. OCT findings vary depending on the stage of disease. In the vitelliform phase, the vitelliform lesion appears on OCT as the accumulation of hyper-reflective material on the apical surface of the RPE in the subretinal space. As the vitelliform lesion gravitates inferiorly, there may be an optically empty, hyporeflective space superiorly with hyperreflective material inferiorly. As the vitelliform lesion is absorbed, OCT shows heterogenous hyper-reflective subretinal material with hyper-reflective clumps that may migrate into the inner retinal layers.
After absorption of the vitelliform material, OCT shows thinning of the ONL and atrophy of EZ and RPE that appear on OCT as hyperreflective columns of light that penetrate to the choroid. In some cases, after the vitelliform material has been reabsorbed, a localized serous detachment remains with an appearance similar to chronic central serous chorioretinopathy (CSCR), with heterogenous yellow subretinal clumps at the border of the serous detachment.7
FAF findings include variable hyper-autofluorescence which correlates with the presence of subretinal vitelliform material and the abnormal accumulation of lipofuscin within RPE cells. In advanced disease, RPE atrophy appears on FAF as hypo-autofluorescent macular lesions. IVFA demonstrates variable early and late hyperfluorescence, which may confound its use in diagnosing leakage due to choroidal neovascularization. OCT-A may be helpful to visualize new vessel growth.
Genetic testing can be a valuable tool for the identification of patients with Best’s disease, especially in the early stages. Although there are no currently available therapies for this condition—aside from treatment with anti-VEGF in cases with choroidal neovascularization—genetic testing allows for the identification of patients for gene therapies or clinical trials.
Pattern Dystrophy
The term “pattern dystrophy” is used to describe a non-specific group of degenerative disorders of the RPE characterized by the deposition of white-yellow material and grayish pigmentary changes at the level of the RPE and photoreceptor outer segments.10
Five distinct phenotypes have been grouped into the category of “pattern dystrophies,” including butterfly pattern dystrophy, adult-onset vitelliform dystrophy (AOFVD), multifocal pattern dystrophy simulating Stargardt’s disease, fundus pulverulentus and reticular dystrophy of the pigment epithelium.10,11 AOFVD is the most common pattern dystrophy.
 |
|
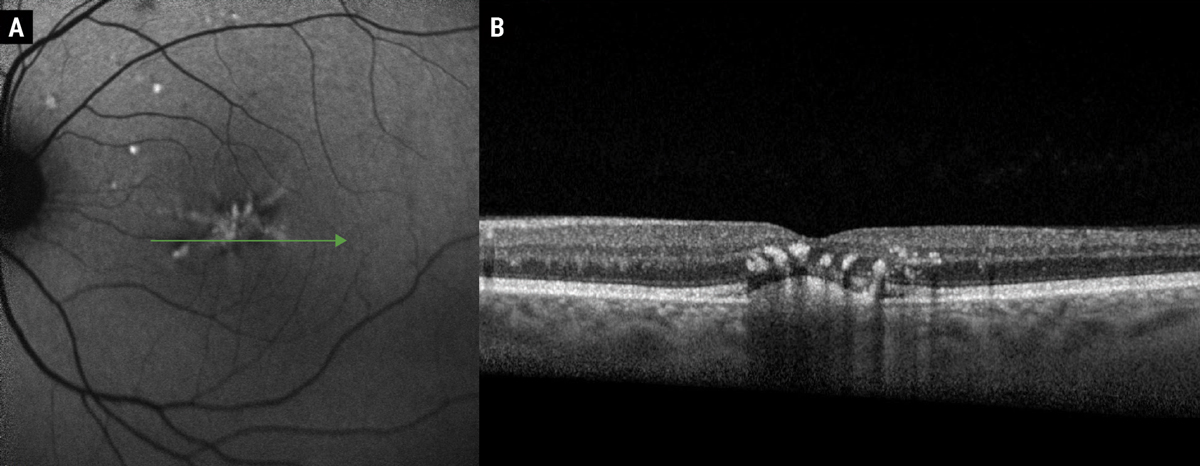
Fig. 3. FAF of butterfly pattern dystrophy (A). Note the hyper-autofluorescent signal in a branching pattern corresponding with subretinal hyper-reflective deposits and accumulation of lipofuscin within RPE cells. OCT (B) demonstrates a subfoveal hyper-reflective subretinal deposit with overlying hyper-reflective foci, representing pigmentary migration. Clinically, these hyperreflective foci create the grayish pigmentation associated with pattern dystrophies. Photos: Mohammad Rafieetary, OD. Click image to enlarge. |
These disorders have historically been considered autosomal dominantly inherited conditions associated with variations in the PRPH2/RDS gene (chromosome 6) which codes for the peripherin protein expressed in photoreceptor outer segments.12 Variations in PRPH2 are thought to result in aberrant metabolism of photoreceptor debris and RPE dysfunction.10,13 Variations in the BEST1 gene (chromosome 1) have also been implicated in pattern dystrophy. BEST1 codes for a calcium chloride transporter found on the basolateral membrane of RPE cells and is linked to other macular dystrophies, including Best’s disease. Additionally linked genes include ABCA4, IMPG1 and CTNNA1.10 Although pattern dystrophies are autosomal dominantly inherited, there is low penetrance, meaning many with these genetic variations do not develop disease.
Individuals with pattern dystrophies may remain asymptomatic until the fifth decade, after which the most common symptoms include mild loss of visual acuity and metamorphopsia.13 Although many individuals retain good vision, as many as 50% of individuals may experience significant loss of central vision due to outer retinal atrophy or choroidal neovascularization around the seventh decade of life.13
Using a combination of imaging modalities, such as OCT, FAF and IVFA, highlights features that help distinguish these conditions from other phenocopies and masqueraders, like AMD, which may become more difficult in advanced disease. Clinically, butterfly pattern dystrophy appears as white-yellow or grayish pigmentary variations with branching arms, resembling a butterfly (Figure 3).10,14 Multifocal pattern dystrophy simulating Stargardt’s presents as scattered pisciform flecks throughout the posterior pole with or without atrophic macular lesions, but without the classic “dark choroid” on IVFA associated with Stargardt’s disease.10,15
AOFVD resembles Best’s disease, though it may be differentiated by age of onset (30 to 50 vs. juvenile onset). Findings include bilateral subfoveal vitelliform lesions with or without a centrally hyperpigmented spot. Vitelliform lesions in AOFVD are typically smaller than vitelliform lesions in Best’s disease (one-third disc diameter vs. one to two disc diameters). Additionally, the EOG Arden ratio in AOFVD is normal to slightly subnormal, while the Arden ratio in Best’s disease is markedly subnormal. AOVFD is associated with a better visual prognosis than Best’s disease, as reading vision is typically preserved. Vision in AOFVD may be compromised by the development of foveal atrophy or CNV.
OCT findings include thickened RPE with hyper-reflective deposits at the level of the RPE or subretinal space. Hyper-reflective foci within the outer retinal layers represent pigmentary migration, which appears clinically as focal areas of grayish hyperpigmentation. Drusen, lipofuscin-rich deposits at the level of Bruch’s membrane, are a hallmark of AMD but not a characteristic finding seen in pattern dystrophies.
Characteristic FAF findings include hyper-autofluorescence corresponding to lipofuscin-rich subretinal deposits. In advanced stages of macular atrophy, FAF demonstrates hypo-autofluorescence in the area of RPE atrophy.
Choroideremia
This disease—an X-linked entity linked to CHM gene variations—affects around one in 50,000 male individuals.16 Specifically, the mutation alters Rab escort protein 1 (REP1), which then manifests as retinal and choroidal atrophy.17 Starting with nyctalopia in the second decade, choroideremia progresses to visual field constriction, with central vision loss occurring decades later.18 Peripheral vision loss leads to a legal blindness diagnosis in the fourth or fifth decade, though some may retain decent central acuity.16,17 Female carriers, however, present differently with minimal visual symptoms and clinical findings of patchy chorioretinal degeneration.19
Early in the disease, peripheral pigmentary changes precede areas of chorioretinal atrophy. These appear as patches of hypoautofluorescence that progress in size and later invade the posterior pole.20 Visual acuity decline coincides with central macular thinning. OCT characteristics include decreased RPE reflectance, ellipsoid zone and external limiting membrane alterations, and outer retinal tubules.20 Inner-layer microcysts signal a negative prognosis and occur in about 20% of cases.20
Choroideremia is incurable, and management options remain supportive in nature or include a referral for low vision services. The adeno-associated virus serotype 2 (AAV2) vector-based gene therapy timrepigene emparvovec—which restores REP1 expression—failed to meet its primary endpoint, though the researchers noted some improvements.21 The retina is ideal for gene therapies due to its post-mitotic status, immune privilege and low dosing.21
 |
|
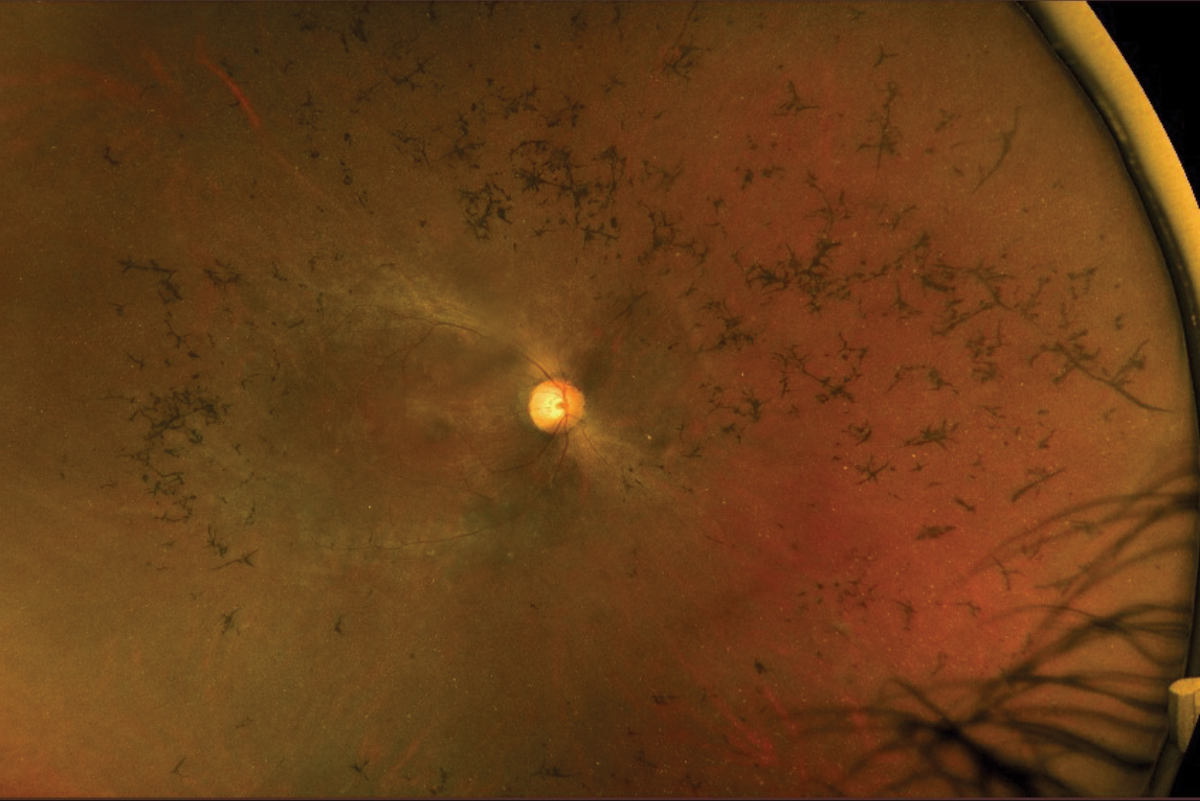
Fig. 4. Fundus photography of an RP patient with mild disc pallor, attenuated arteries, pigmentary changes and bone spicules. Photo: Mohammad Rafieetary, OD. Click image to enlarge. |
Besides offering genetic testing, clinicians stumble when educating choroideremia patients since disease progression from nyctalopia and visual field constriction to eventual vision loss varies widely. The 20-month NIGHT study tackled this question and aimed to provide insight into choroideremia’s natural history. Unlike previous studies that only assessed central acuity, these researchers added functional and anatomical outcomes to see which ones might highlight disease progression. In early disease, BCVA lacked the sensitivity of other measures such as retinal sensitivity, central ellipsoid zone area and total area of FAF.22
Central Areolar Choroidal Dystrophy
This predominately autosomal dominant condition affects the macula and leads to well-demarcated outer retinal, RPE and choroidal atrophy.23,24 In doing so, it acts as a masquerader to geographic atrophy caused by AMD. The difference, however, lies in the symmetrical appearance, lack of drusen and earlier onset (second to fourth decades).23 There is a late-onset variety that appears in the sixth to eighth decades that can confound diagnosis.23 Peripherin/RDS (PRPH2) gene mutation is the most common cause, but it has also been linked to GUCA1A, GUCY2D, CDHR1, ABCA4 and TTLL5.25
This type of dystrophy is a progressive disorder with four recognized stages. Focal parafoveal pigment changes mark stage 1, which later develops into an oval shaped area of macular atrophy in stage 2.23 Well-demarcated RPE atrophy with subsequent foveal atrophy represent stages 3 and 4, respectively.25 Visual acuity generally declines in the later stages.
Electroretinogram (ERG) and EOG help distinguish this from other macular pathologies, as they are both normal in most cases.24 FAF may reveal speckled hyper- and hypo-autofluorescence in the early stages, with atrophic areas appearing as solely hypo-autofluorescent areas in the late stages. OCT depicts EZ disruption early in the disease, with subsequent RPE and choroidal atrophy later in the process. No treatment exists for the condition.
Leber’s Congenital Amaurosis (LCA)
While this condition accounts for just 5% of all IRDs, LCA is the most severe with the earliest onset, presenting in infancy.26 A less devastating form—early-onset severe retinal degeneration (EOSRD)—presents after infancy and before age five.26 Variants in at least 25 genes cause the vastly autosomal recessive condition, which can occur alone or as part of a syndrome (Senior-Loken or Joubert).27 The most commonly identified genes are GUCY2D, CEP290, CRB1, RDH12 and RPE65.28
Besides severe visual impairment, LCA patients exhibit nystagmus, poor pupil responses and a mostly undetectable full-field ERG.28 Patients are often highly hyperopic. A common occurrence is the oculodigital sign or eye rubbing, which may be an attempt to stimulate a visual signal. Interestingly, keratoconus often accompanies LCA, though it may be due to other genetic factors vs. oculodigital interactions.29
Clinically, the fundus may appear unremarkable or show signs of RPE mottling. Later presentations vary and include nummular or bone spicule pigmentation, salt and pepper retinopathy, vessel attenuation, macular atrophy and optic disc pallor.27 Almost 90% of RPE65 mutations showed an absent or severely diminished FAF.30
Management is targeted at symptoms. The rate of vision loss varies, with faster progression noted in some genes.31 Some LCA patients may exhibit speech, social skill and behavioral problems, necessitating a multidisciplinary approach in their care.31 A treatment option does exist, however, only for those inflicted with a biallelic RPE65 variant.
In 2017, the FDA approved Luxturna as the first, and still only, retinal gene therapy. The costly, one-time subretinal injection of voretigene neparvovec-rzyl contains a healthy copy of the gene. The RPE65 mutation accounts for only 5% to 10% of LCA cases as well as 2% of RP cases, highlighting the importance of genetic testing in individuals with these conditions.27
 |
|
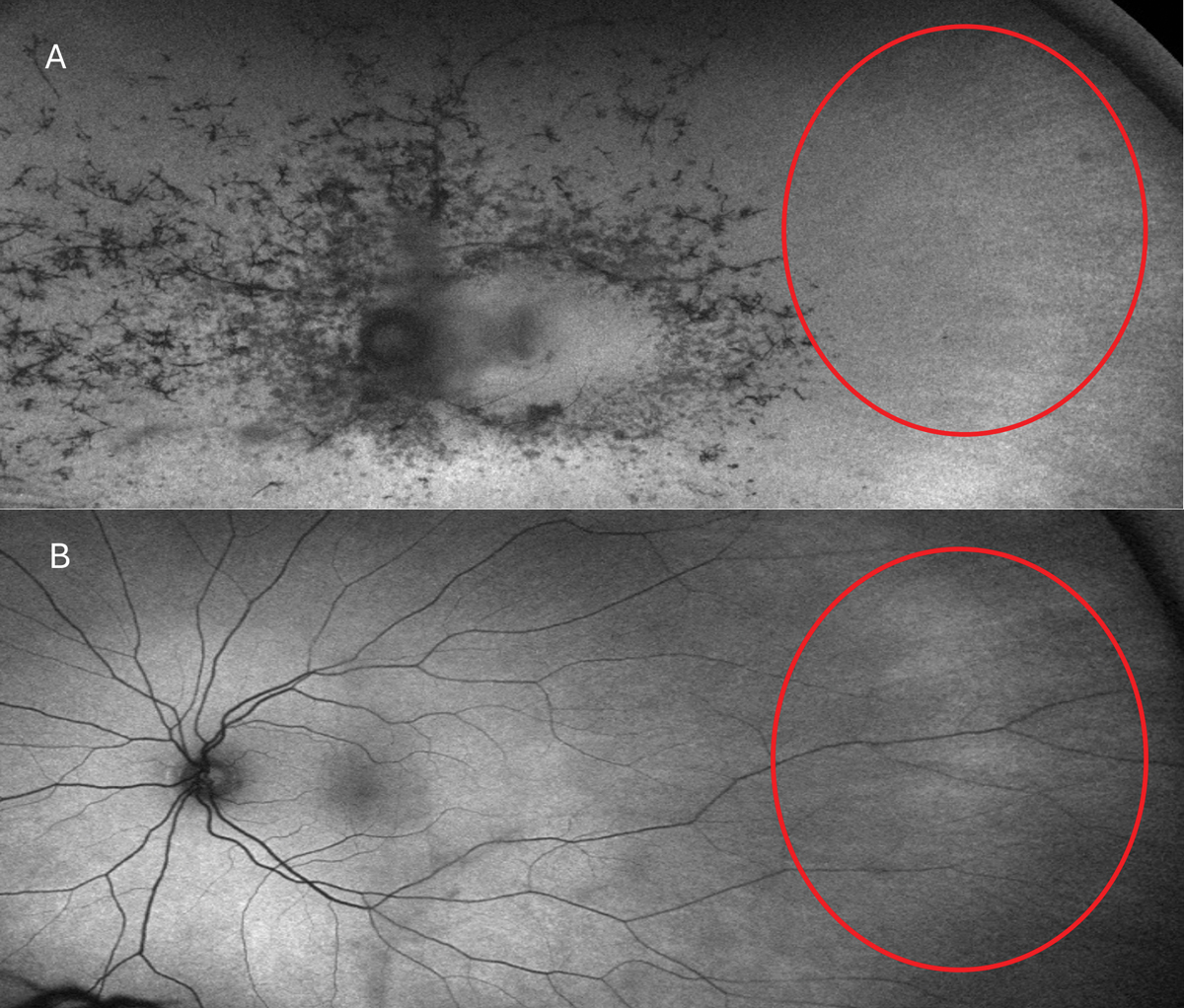
Fig. 5. Typical RP FAF presentation (A). In the red circles, note the attenuation and absence of retinal arteries vs. that of an RP carrier (B). Photo: Mohammad Rafieetary, OD.Click image to enlarge. |
Retinitis Pigmentosa
The most common inherited retinal disease, RP is a heterogeneous group of IRDs characterized by progressive loss of rod and cone photoreceptors, leading to visual impairment. It has a variable prevalence globally, affecting around one in 4,000 in the US and one in 5,000 individuals worldwide.32
RP is primarily caused by genetic variants affecting photoreceptors or RPE cells.33 More than 100 genetic loci on at least 40 different genes have been identified in patterns of inheritance and expression for RP, and it is likely that more have yet to be discovered.34,35 The pattern of inheritance of these genes can be autosomal dominant (AD), autosomal recessive (AR), X-linked recessive or dominant (affected X chromosome on both parents sides), as well as genetic mutation. In some cases, the mode of inheritance remains unknown.36
Common symptoms include progressive vision loss, night blindness or nyctalopia, an enlarged blind spot leading to peripheral vision loss and eventual central vision loss, posing challenges for tasks like reading, driving or facial recognition.37 The symptoms of RP can vary greatly in terms of onset, severity and progression, even among family members affected by the condition, due to variable genotypic penetrance. Epigenetic factors and potentially environmental influences are thought to play a role in this variability, making it difficult to establish genotype-phenotype correlations.38
The classic clinical signs of RP observed during fundus examinations include a pale optic disc, retinal vessel attenuation and “bone-spicule” hyperpigmentation (Figures 4 and 5). These retinal changes typically occur bilaterally and exhibit a high degree of symmetry. Other fundus findings include posterior capsular cataracts, optic nerve drusen, cystoid macular edema (CME) and inner retina cystic atrophy, epiretinal membrane formation and Coats-like disease—a mid-peripheral exudative vasculopathy characterized by telangiectatic vessels, focal serous retinal detachment and lipid exudate deposition.39,41 The onset and presentation of these findings vary widely among individuals and may even appear in atypical forms such as a unilateral or sectoral RP presentation.40
Retinitis pigmentosa can be categorized into non-syndromic (70% to 80% of cases) and syndromic (20% to 30% of cases) forms based on the presence of systemic abnormalities and other associated diseases. Non-syndromic RP has no systemic abnormalities, whereas syndromic RP is accompanied by non-ocular syndromes and systemic disease.37
Usher syndrome, a syndromic form of RP, encompasses a group of genetic disorders classified into three subtypes: Usher syndrome type I (USH1), type II (USH2) and type III (USH3). These are based on specific characteristics, such as the presence of vestibular involvement, the age of RP onset and the rate of disease progression. Numerous genes are implicated in this syndrome, contributing to its genetic heterogeneity.
Usher syndrome affects up to one in 6,000 individuals and is the leading cause of deafblindness in humans. Symptoms typically manifest from birth (in USH1 and USH2) or later in mid-childhood or adulthood (USH3). Common symptoms include congenital hearing loss, loss of night vision as the initial visual symptom and the development of blind spots leading to progressive peripheral vision loss.
A diagnosis of USH1 is considered when a patient presents with congenital profound bilateral sensorineural hearing loss, often accompanied by severe vestibular abnormalities that may not be clinically obvious. Early-onset RP, which progresses slowly, is another hallmark feature. Affected individuals typically exhibit abnormal speech development and vestibular areflexia, characterized by a lack of normal vestibular reflexes, a defining trait of USH1. Consequently, children with USH1 often experience delayed walking compared to their peers due to vestibular impairment. Balance issues may persist into adulthood, while the remainder of the physical examination typically appears normal.
USH2 is characterized by congenital bilateral sensorineural hearing loss that primarily affects higher frequencies, along with intact vestibular function. Unlike USH1, RP onset in USH2 typically occurs during adolescence or adulthood. This subtype represents the most common form of Usher syndrome, accounting for 75% to 80% of diagnosed cases.
USH3 is distinguished by post-lingual progressive sensorineural hearing loss, meaning the hearing loss develops after speech has been acquired and worsens over time. RP onset in USH3 typically occurs later compared to other types of Usher syndrome. Vestibular function can vary in USH3, with affected individuals experiencing impairment in various degrees. In USH2 and USH3, the progression of RP is more noticeable, likely due to its later onset compared to USH1.
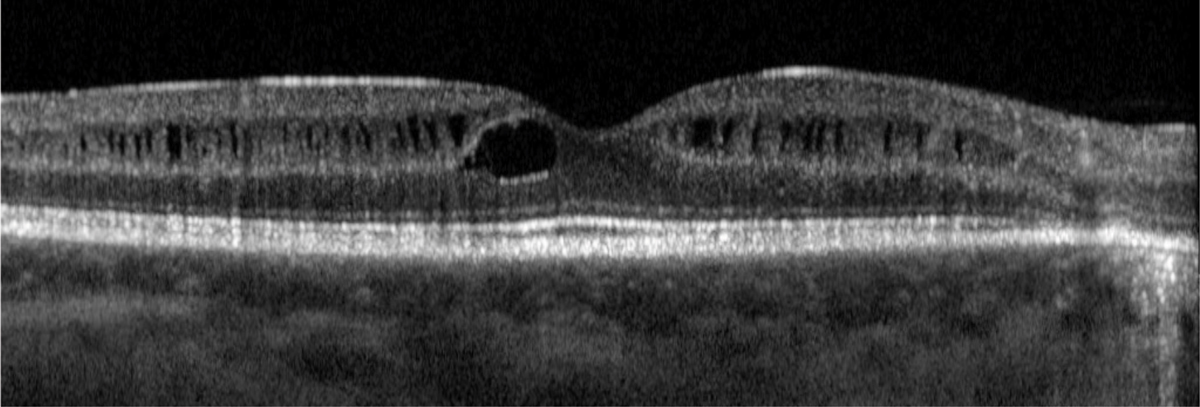
In addition to the typical features of Usher syndrome, there are some uncommon ocular manifestations associated with this condition. Intraretinal cystoid spaces have been observed, particularly in individuals with USH2 (Figure 6). Additionally, rare instances of bilateral Coats-like exudative retinopathy have been reported in Usher syndrome, indicating abnormal blood vessel growth and leakage in the retina resembling Coats disease.
ERG is considered the gold standard for diagnosing RP, establishing baseline function and monitoring disease progression. ERG can detect photoreceptor dysfunction even when changes on a clinical exam or imaging are minimal. While ERG remains the standard of care for diagnosis, FAF can be used instead of ERG to monitor disease progression, particularly in later stages when ERG may be less reliable.
 |
|
Fig. 6. Intraretinal cystoid spaces have been described in patients with Usher syndrome. This is more commonly seen in USH2. Photo: Mohammad Rafieetary, OD. Click image to enlarge. |
Visual field testing is also valuable for establishing baseline function and monitoring disease progression. In the early stages of RP, visual field measurements demonstrate variable peripheral vision loss, progressing to a ring scotoma consistent with the tunnel vision described in later disease stages.
OCT can be used to evaluate retinal morphological changes in RP patients. In the early stages of the disease, OCT can reveal disorganization of the outer retinal layers. As RP progresses, there is a noticeable decrease in the thickness of the outer nuclear layer. In the advanced stages of RP, complete loss of both the outer segment and the outer nuclear layer occurs, while the inner retinal layers remain relatively well preserved.41
Cone-Rod Dystrophy
This condition is another IRD that typically manifests in childhood and progresses over time, affecting approximately one in 30,000 individuals. It is frequently mistaken for RP but differs by predominantly affecting cones over rods. At least 10 genes have been found associated with cone-rod dystrophy, which is usually passed on in an AD pattern. The mutation in the GUCY2D and CRX genes account for 50% of cases.
Patients initially experience vision loss and color vision abnormalities, followed by peripheral field constriction. Fundus examination in the early stages shows macular pigmentation and atrophy, progressing to peripheral bone spicule pigmentation in advanced cases, often affecting the mid-periphery later in the disease.
Common symptoms include decreased visual acuity, light sensitivity, difficulty recognizing colors, blind spots in the visual field and a gradual loss of peripheral vision leading to blindness by mid-adulthood. Diagnosis of cone-rod dystrophy relies on ERG changes, indicating more severe cone impairment compared to rods.42
Takeaways
The value of clinical examination and ancillary testing should remain paramount when evaluating IRDs. The American Academy of Ophthalmology recommends that the clinical evaluation of suspected IRDs should involve a comprehensive examination combined with imaging, including color fundus photos, FAF, OCT, visual field testing, full-field ERG and genetic testing.1
Diagnosing inherited retinal diseases can be challenging due to overlapping symptoms. Therefore, genetic testing is an essential tool to identify the underlying genetic cause of vision loss or impairment. In cases of suspicion for IRD, genetic testing should be pursued with the caveat that current testing platforms are only 60% to 70% successful in identifying the causative gene. Patients who undergo genetic testing should then be referred for genetic counseling.41
When considering genetic testing, note that several groups offer it at no cost. Clinicians should be familiar with the process and the paperwork. The Foundation Fighting Blindness partners with Blueprint Genetics to make available the My Retina Tracker panel, while Invitae has the sponsored Inherited Retinal Disorders Panel, although it was recently purchased by LabCorp, so stay tuned for any future changes. Most importantly, both provide genetic counseling to help interpret the results and discuss how they affect the patient now and in the future.
Patients should sign up for the My Retina Tracker Registry—a research database of the Foundation Fighting Blindness. The registry is designed to share information about rare retinal diseases, including IRDs, to identify individuals who might be interested in participating in research studies and clinical trials.
Finally, consider a low vision consult, even if the patient is asymptomatic, as these specialists would welcome the chance to introduce themselves and begin early conversations.
Dr. Attar is an assistant professor in the department of ophthalmology and director of optometric services at the University of Mississippi Medical Center. She is a fellow of the American Academy of Optometry (AAO), chair of the AAO Retina Special Interest Group and vice chair of the American Optometric Society Leadership Development Committee. She is on advisory boards for Heidelberg Engineering, Apellis and OcuTerra Therapeutics.
Dr. Steele completed her ocular disease residency at the Memphis VA. She is currently completing her fellowship at the Charles Retina Institute and is a consulting faculty member at Southern College of Optometry. She has no financial disclosures.
Dr. Williamson is the residency supervisor at the Memphis VA Medical Center. He holds faculty positions at multiple optometry schools and allied health programs and is a fellow of the American Academy of Optometry and the Optometric Retina Society. Dr. Williamson also serves as the secretary for the Optometric Retina Society. He has no financial disclosures.
1. Duncan JL, Branham K, Birch DG, et al. Guidelines on clinical assessment of patients with inherited retinal degenerations – 2022. American Academy of Ophthalmology. www.aao.org/education/clinical-statement/guidelines-on-clinical-assessment-of-patients-with. October 2022. Accessed April 15, 2024. 2. Al-Khuzaei S, Broadgate S, Foster CR, et al. An overview of the genetics of ABCA4 retinopathies, an evolving story. Genes (Basel). 2021;12(8):1241. 3. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101(1):25-30. 4. Li CHZ, Pas JAAH, Corradi Z, et al. Study of late-onset Stargardt type 1 disease: Characteristics, genetics and progression. Ophthalmology. 2024;131(1):87-97. 5. Lambertus S, van Huet RA, Bax NM, et al. Early-onset Stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015;122(2):335-44. 6. Heath Jeffery RC, Chen FK. Stargardt disease: Multimodal imaging: a review. Clin Exp Ophthalmol. 2021;49(5):498-515. 7. Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmology. 2006;113(8):1392-1400. 8. Dalvin LA, Pulido JS, Marmorstein AD. Vitelliform dystrophies: prevalence in Olmsted County, Minnesota, United States. Ophthalmic Genet. 2017;38(2):143-7. 9. Querques G, Zerbib J, Santacroce R, et al. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol Vis. 2009;15:2960-72. 10. Hanif AM, Yan J, Jain N. Pattern dystrophy: an imprecise diagnosis in the age of precision medicine. Int Ophthalmol Clin. 2019;59(1):173-94. 11. Gass JDM. Stereoscopic atlas of macular diseases: Diagnosis and treatment. Mosby. 1997. 12. Conley SM, Naash MI. Gene therapy for PRPH2-associated ocular disease: challenges and prospects. Cold Spring Harb Perspect Med. 2014;4(11):a017376. 13. Sodi A, Mucciolo DP, Giorgio D, et al. Clinical and molecular findings in patients with pattern dystrophy. Ophthalmic Genet. 2021;42(5):577-87. 14. Kumar V, Kumawat D. Multimodal imaging in a case of butterfly pattern dystrophy of retinal pigment epithelium. Int Ophthalmol. 2018;38(2):775-9. 15. Rotsos T, Gkounta A, Symeonidis C, et al. Multifocal pattern dystrophy simulating fundus flavimaculatus: Multimodal imaging for early diagnosis. Case Rep Ophthalmol. 2021;12(2):724-8. 16. Khan K, Islam F, Moore A, et al. Clinical and genetic features of choroideremia in childhood. Ophthalmology. 2016;123(10):2158-65. 17. Jauregui R, Park K, Tanaka A, et al. Spectrum of disease severity and phenotype in choroideremia carriers. Am J Ophthalmol. 2019;207:77-86. 18. Aleman T, Han G, Serrano L, et al. Natural history of the central structural abnormalities in choroideremia: a prospective cross-sectional study. Ophthalmology. 2017;124(3):359-73. 19. MacDonald IM, Russell L, Chan C. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol. 2009;54(3):401-7. 20. Brambati M, Borrelli E, Sacconi R, et al. Choroideremia: update on clinical features and emerging treatments. Clin Ophthalmol. 2019;13:2225-31. 21. MacLaren R, Fischer M, Gow J, et al. Subretinal timrepigene emparvovec in adult men with choroideremia: a randomized Phase III trial. Nat Med. 2023;29(10):2464-72. 22. MacLaren R, Lam B, Fischer D, et al. A prospective, observational, non-interventional clinical study of participants with choroideremia: The NIGHT study. Am J Ophthalmol. 2024;263:35-49. 23. Paez-Escamilla M, Jhingan M, Gallagher D, et al. Age-related macular degeneration masqueraders: from the obvious to the obscure. Surv Ophthalmol. 2021;66(2):153-82. 24. Camargo J, Prezia G, Shiokawa N, et al. New insights on the regulatory gene network disturbed in central areolar choroidal dystrophy – beyond classical gene candidates. Front Genet. 2022;13:886461. 25. Smailhodzic D, Fleckenstein M, Theelan T, et al. Central areolar choroidal dystrophy (CACD) and age-related macular degeneration (AMD): differentiating characteristic in multimodal imaging. Invest Ophthalmol Vis Sci. 2011;52(12):8908-18. 26. Kondkar A, Abu-Amero K. Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. Exp Eye Res. 2019;189:107834. 27. Varela M, Guimaraes T, Georgiou M, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Current management and clinical trials. Br J Ophthalmol. 2022;106(4):445-51. 28. Kumaran N, Moore A, Weleber R, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147-54. 29. Elder MJ. Leber congenital amaurosis and its association with keratoconus and keratoglobus. J Pediatr Ophthalmol Strabismus. 1994;31(1):38-40. 30. Chung D, Bertelsen M, Lorenz B, et al. The natural history of inherited retinal dystrophy due to biallelic mutations in the RPE65 gene. Am J Ophthalmol. 2019;199:58-70. 31. Georgiou M, Fujinami K, Michaelides M. Inherited retinal diseases: Therapeutics, clinical trials and end points – a review. Clin Experiment Ophthalmol. 2021;49(3):270-88. 32. O’Neal TB, Luther EE. Retinitis pigmentosa. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. www.ncbi.nlm.nih.gov/books/NBK519518. Updated February 19, 2023. Accesed April 15, 2024. 33. Yang YJ, Peng J, Ying D, Peng QH. A brief review on the pathological role of decreased blood flow affected in retinitis pigmentosa. J Ophthalmol. 2018;2018:3249064. 34. Berson EL. Retinitis pigmentosa and allied diseases. In: Albert D, Miller J, Azar D, Blodi B, eds. Albert and Jakobiec’s Principles and Practice of Ophthalmology, 3rd ed. Philadelphia, PA: Elsevier; 2008. 35. Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238-49. 36. Bravo-Gil N, González-Del Pozo M, Martín-Sánchez M, et al. Unravelling the genetic basis of simplex retinitis pigmentosa cases. Sci Rep. 2017;7:41937. 37. Cross N, van Steen C, Zegaoui Y, et al. Retinitis pigmentosa: Burden of disease and current unmet needs. Clin Ophthalmol. 2022;16:1993-2010. 38. Dvoriantchikova G, Lypka KR, Ivanov D. The potential role of epigenetic mechanisms in the development of retinitis pigmentosa and related photoreceptor dystrophies. Front Genet. 2022;13:827274. 39. Ruff A, Tezel A, Tezel TH. Anatomical and functional correlates of cystic macular edema in retinitis pigmentosa. PLoS One. 2022;17(10):e0276629. 40. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157-86. 41. Nguyen XT, Moekotte L, Plomp AS, et al. Retinitis pigmentosa: Current clinical management and emerging therapies. Int J Mol Sci. 2023;24(8):7481. 42. Chawla H, Vohra V. Retinal dystrophies. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. www.ncbi.nlm.nih.gov/books/NBK564379. Updated March 16, 2023. Accessed April 15, 2024. |