A Stepwise Approach to Evaluating Diplopia and Other Ocular Motility Abnormalities
An optometrist can carefully whittle down the root of patients’ ocular misalignments by taking a following a specific order of operations.
By Kelly A. Malloy, OD, Erin M. Draper, OD, Ashley K. Maglione, OD, and Kelsey L. Moody, OD
Release Date:
May 2016Expiration Date:
May 1, 2019Goal Statement:
Issues of ocular motility or diplopia can be symptoms of a broad range of neuro-ophthalmic conditions. To narrow down the suspects, optometrists should follow a particular process of elimination based on the patient’s symptoms as well as brain scanning technology. This article provides a detailed overview of that process and defines conditions optometrists may encounter.Faculty/Editorial Board:
Kelly A. Malloy, OD, Erin M. Draper, OD, Ashley K. Maglione, OD, and Kelsey L. Moody, ODCredit Statement:
This course is COPE approved for 2 hours of CE credit. COPE ID is 49753-NO. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.Joint-Sponsorship Statement:
This continuing education course is joint-sponsored by the Pennsylvania College of Optometry.Disclosure Statement:
The authors have no relationships to disclose.When patients present with ocular misalignment or diplopia, the number of possible etiologies can be overwhelming. It requires a systematic, stepwise approach to both examination techniques and differential diagnoses to attain a greater level of comfort with these patients.
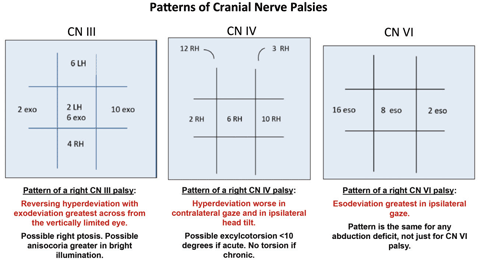 |
| Click image to enlarge. |
To establish this stepwise approach, first review some basic anatomy. This will help localize the problem. Once acheived, you can narrow in on a particular anatomical structure and evaluate for known etiologies to that region.
For a stepwise approach to identifying the cause of diplopia, think of four distinct categories. First, consider if the findings localize to the brain, including the cerebral cortex and brainstem. Second, consider whether the findings localize to a specific cranial nerve (CN) or a combination of nerves and if a specific location along the CN course can be pinpointed. Third, evaluate for features that suggest localization to the neuromuscular junction. Fourth, look for suggestions of localization to the orbit. Evaluating diplopia or ocular misalignment in this manner ensures all possible options for the clinical presentation are considered.
A careful examination of the efferent visual system is necessary in all patients with diplopia or ocular misalignment. For more information on the basics of performing the efferent visual system evaluation, refer to the February 2015 Review of Optometry article on that topic.1
This article provides an anatomical review of the structures key to ocular motility followed by four main sections, each dedicated to a specific anatomic location (brain, nerve, junction, orbit). Each of these sections discuss clinical features that may be present at that anatomic location, potential abnormalities that may occur there and subsequent work-up needed to make a diagnosis.
Anatomy Review
Recall there is cortical, or supranuclear, input from several areas, including the frontal and parietal lobes of the brain. The brainstem plays a major role in ocular motilities, housing not only CNs III, IV and VI nuclei and beginning segments of each corresponding CN, but also important anatomic regions that coordinate movements between eyes. Understand the pathway that each CN takes after it exits the brainstem on its way to the orbit to innervate specific extraocular muscles (EOMs), as these nerves ultimately control eye movements. For the EOMs to receive signals from their corresponding CNs, the neuromuscular junction (NMJ) must be intact and functioning properly.
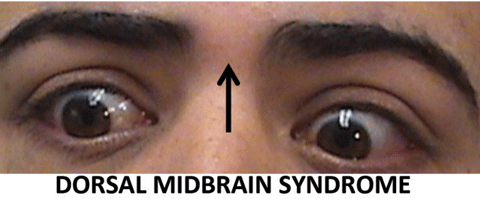
 |
| These images portray patients displaying brainstem motility disorders. Click image above to enlarge. |
 |
With so many components working together for normal ocular motility, it can be a daunting task to determine the location of the system’s breakdown. However, if approached in a stepwise fashion, it is less overwhelming.
First, assess the patient’s ability to perform ductions. Do this by having the fellow eye occluded and looking only at one eye, assessing in which positions of gaze the eye is not able to achieve full range of motion. Next, carefully perform cover testing in all positions of gaze to identify whether the deviation is comitant or non-comitant, and to look for any classic pattern suggesting a CN palsy or other identifiable process. Signs or symptoms can help narrow your focus, but familiarity with neuroanatomy is key to diagnosis here.
EOM Disorders Stemming From the Brain
When patients present with EOM issues, the clinician should first evaluate them for disorders related the brain, since this is where the efferent pathways begin. Here is a list of likely diagnoses and how to identify them.
Gaze palsy—If the patient experiences an inability to look to one side, as well as weakness of the lower face and arms or legs on the same side, they may be suffering from a gaze palsy. This is a reduced ability of both eyes to move in the direction of lateral gaze.
The frontal eye fields are regions located anterior to the motor areas in the cerebral cortex. Their function allows contralateral gaze. Damage to these fields results in contralateral gaze palsy, and possibly ipsilateral gaze preference.2,3 Because of their proximity to the motor cortex, these lesions in the frontal eye fields could also cause contralateral weakness.
An intact cerebral cortex communicates through the contralateral paramedian pontine reticular formation (PPRF) with the contralateral CN VI nucleus in the pons. This is the horizontal gaze center, coordinating both eyes in ipsilateral gaze. The contralateral CN VI nucleus gives rise to two populations of fibers. One becomes CN VI, innervating its ipsilateral lateral rectus muscle, and the other travels through an interneuron to the contralateral medial longitudinal fasciculus (MLF), the subnucleus of CN III, and ultimately the medial rectus muscle. A lesion of the CN VI nucleus results in ipsilateral gaze palsy. Pontine lesions large enough to also affect the ventral motor tracts could cause limb weakness both contralateral to the lesion and to the direction of gaze palsy.
This differs from cortical lesions, where weakness and gaze palsy are on the same side. Another difference is that nuclear lesions cannot be overcome with doll’s head technique or vestibulo-ocular reflex (VOR), whereas supranuclear lesions can.3,4
Abduction deficit (CN VI damage in the pons)—Damage to CN VI in the pons (sparing VI nucleus) produces a CN VI palsy, or abduction deficit. These deficits are not specific to CN VI palsy; they can occur at all four anatomic regions. An abduction deficit with associated ipsilateral facial weakness, contralateral extremity weakness or reduced contralateral sensation significantly increases suspicion for a pontine lesion. Look for facial asymmetry; ask patients to raise their eyebrows, frown, puff out their cheeks and smile. As CN VI travels through the pons, dorsal damage near the sensory fibers (medial lemniscus) causes decreased contralateral sensation of the limbs due to sensory decussation in the medulla.5,6 Ventrally, CN VI can be damaged near the motor fibers, resulting in contralateral extremity weakness.5,6
Internuclear ophthalmoplegia—When the medial longitudinal fasciculus (MLF) is damaged in the pons or midbrain, the medial rectus does not get the signal from the horizontal gaze center of the contralateral VI nucleus.
 |
| When CN VI does not develop properly in utero, the lateral rectus gets innervated by CN III, leading to a congenital cranial dysinnervation disorder called Duane’s retraction syndrome. It can present as unilateral or, as in this case, bilateral. Click image to enlarge. |
The result is an adduction deficit and exo deviation increasing in the direction of attempted lateral gaze. Associated abducting nystagmus in the fellow eye is pathognomonic for internuclear ophthalmoplegia (INO). To further localize INO to the pons or midbrain, assess the convergence. Convergence is spared in pontine lesions; convergence may be affected in midbrain lesions if the medial rectus subnuclei of CN III are also affected.7
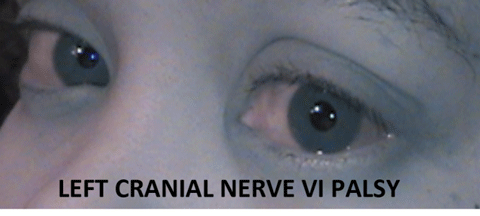 |
| This patient was determined to have a left CN VI palsy only after complete work-up. Without knowing any other information, this abduction deficit could be associated with any of the four locations: brain, nerve, neuromuscular junction or orbit. A detailed clinical examination can help sort this out. |
One and a half syndrome—A lesion affecting CN VI nucleus (or PPRF) and ipsilateral MLF results in an ipsilateral gaze palsy and INO. The latter is named for the eye with the adduction deficit. A lesion of the right CN VI nucleus and the right MLF results in a right gaze palsy and right INO.7
Duane’s retraction syndrome—When CN VI does not develop properly in utero, the lateral rectus gets innervated by CN III and the patient has an ipsilateral abduction deficit. In contralateral gaze—because of co-contraction of medial and lateral recti—the globe is pulled back in the orbit, giving an enophthalmic appearance and decreased palpebral aperture, pathognomonic for type I Duane’s retraction syndrome. This congenital cranial dysinnervation disorder may be unilateral or bilateral and does not require workup or treatment.8-10
CN IV damage in the brainstem—The CN IV nucleus is located in the lower dorsal midbrain. Unlike other CNs, CN IV has a short course in the brainstem because it exits posteriorly; it is not associated with contralateral weakness or numbness. After CN IV exits the midbrain, it crosses in the isthmus pons. Damage to the anterior medullary velum, where both CN IV cross, can result in bilateral CN IV palsy.5,6
Brainstem CN palsy and Horner’s syndrome—With any CN palsy from a brainstem lesion, there may be an associated Horner’s syndrome, due to sympathetic involvement. With CN VI and CN III involvement, Horner’s syndrome only presents ipsilateral to the palsy, since the sympathetic fibers do not cross. With CN IV involvement, Horner’s syndrome is contralateral.
Dorsal midbrain syndrome—Damage to the posterior commissure, results in pathognomonic features, including difficulty with upgaze, eyelid retraction, light-near dissociation pupils and convergence retraction nystagmus. With lesions also affecting surrounding anatomic areas, unilateral or bilateral CN IV palsy or skew deviation may also be present.3,11,12
Skew deviation—This phenomenon is a vertical misalignment of the eyes, with the higher eye intorted and lower eye extorted; a head tilt is often seen toward the lower eye. This occurs with damage to the pathway connecting the vestibular and ocular motor systems. This pathway involves semicircular canals, vestibular nerve and nuclei in lateral medulla and the MLF that interconnects CN IV nucleus and CN III subnuclei to control vertical alignment and torsion.3 Since the MLF also is involved in coordination of horizontal movements, it is not uncommon to see an INO and skew deviation together.
CN III damage in the midbrain—The CN III nucleus, or ocular motor complex, sits midline in the midbrain tegmentum. Nuclear lesions are rare, resulting in bilateral deficits. During its course, CN III can be affected with sensory fibers, resulting in a loss of contralateral sensation. CN III can be affected ventrally with the crus cerebri motor tracts, resulting in contralateral weakness. CN III travels near the red nucleus, which is involved with motor coordination; there may be contralateral ataxia and difficulty with motor control.5,6
If features that localize to the cortex or brainstem are found, referral to neurology or neuro-ophthalmology is warranted. Patients need neuroimaging (preferably MRI with contrast if not contraindicated) to look for infarct, hemorrhage, demyelination, primary tumor or metastatic disease.
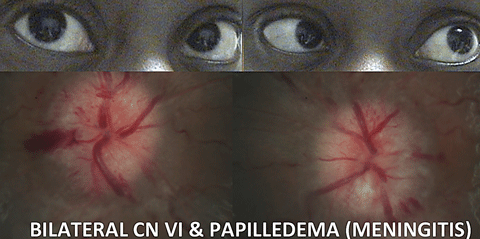 |
| In cases of CN VI, always assess the optic discs for features of even subtle papilledema, since this constitutes a medical emergency. Click image to enlarge. |
Evaluating for EOM Disorders Stemming from the Nerves
Once CNs III, IV and VI leave the brainstem, they travel through the subarachnoid space to the cavernous sinus prior to entering the orbit through the superior orbital fissure. This section follows the nerves along this pathway, where they are subject to damage from various etiologies.
Subarachnoid space—Within the subarachnoid space, the CNs are subject to trauma; CN IV is most likely to incur damage, due to its thin caliber, long course from the dorsal aspect of the brainstem and position in the tentorial margin.6 Always ask patients about their trauma history.
Additionally, within the subarachnoid space, each CN may be affected by a subarachnoid hemorrhage, compressive mass, cerebral spinal fluid (CSF) inflammation (i.e., sarcoidosis) or CSF infection (i.e., meningitis, Lyme disease, syphilis and fungal).5,17-19 Any of these processes could result in increased intracranial pressure (ICP). CN VI is most affected by increased ICP. One or both CN VI may be compressed by the petroclinoid ligament while traveling through Dorello’s canal to the cavernous sinus.20 Always consider increased ICP with an abduction deficit. Carefully assess the optic discs for features of even subtle papilledema, which constitutes a medical emergency. Necessary work-up includes brain MRI and MRV (to rule out cerebral venous sinus thrombosis), followed by a lumbar puncture to measure opening pressure and rule out infectious or inflammatory CSF processes. It is also possible to see a CN VI palsy with a sudden decrease in ICP, such as with a CSF leak after lumbar puncture.
While in the subarachnoid space, CN VI traverses up the clivus in route to the cavernous sinus. Here, one or both CN VI may be subject to damage from bone metastases, most commonly seen in lung, breast or prostate cancers.21 The cisternal and cavernous sinus segments of CNs are also subject to paresis from schwannomas and metastatic spread in CSF.22
Another condition that emerges from the subarachnoid space is aneurysm. Although aneurysms may affect any artery and compress any adjacent CN, CN III is most commonly affected.23 Since the parasympathetic preganglionic fibers travel on the outside of CN III, external compression of CN III results in pupillary dilation in addition to the tell-tale reversing hyper deviation pattern on cover testing. Any painful or pupil-involved CN III palsy is considered an aneurysm of the posterior communicating artery until proven otherwise. Due to the high risk of mortality, emergent work-up is necessary and should include MRA, CTA or formal angiography.
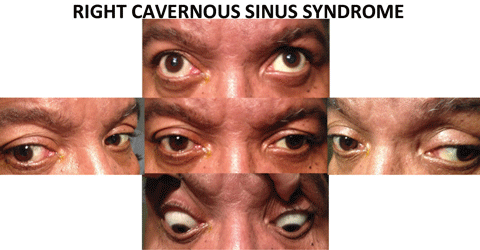 |
| Above, the combination of a right CN III pattern as well as a right abduction deficit, localizes the lesion to the cavernous sinus. These findings may be subtle, which underscores the need for cover testing in all positions of gaze. Also, testing for decreased facial sensation in V1 and V2 also helps with localization. Below, evidence of pupil-involved CN III palsy requires an emergent work up, due to the risk of mortality associated with aneurysm. Click images to enlarge. |
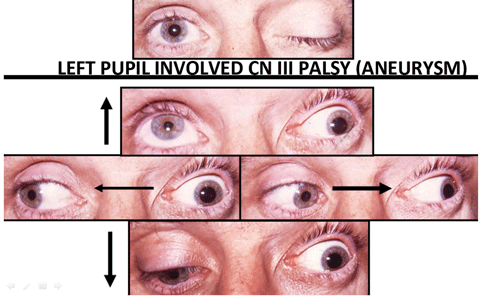 |
Cavernous Sinus—CNs III, IV, and VI converge in the cavernous sinus. With multiple cranial nerve involvement, be suspicious of an orbital apex or cavernous sinus lesion such as neoplasm, carotid artery aneurysm, inflammation, fistula or thrombosis.24
Patients with cavernous sinus syndrome may complain of pain, since the first division of the trigeminal nerve (V1) may be affected. If a cavernous sinus lesion is suspected, test for decreased facial sensation in the regions innervated by CN V1 and CN V2, above and below the orbit respectively. While V1 and V2 travel in the lateral wall of the cavernous sinus, V3 is not in the cavernous sinus. Therefore, the preservation of sensation in the chin region may help confirm localization. Depending on the nature of the lesion, chemosis, proptosis or Horner’s syndrome may also be present.
Neoplasms—Cavernous meningiomas are common lesions of the base of the skull. Although they’re benign, they can be invasive and difficult to treat.25 If lesions extend into the orbital cavity, you may observe optic atrophy. Of course, malignant tumors, such as lymphoma, and even metastases may also result in cavernous sinus syndrome. Be suspicious of a metastatic process in patients with a history of cancer.
The pituitary gland sits between the two cavernous sinuses; it is not uncommon for a pituitary macroadenoma to compress or extend into one or both sinuses. Rarely, a pituitary hemorrhage or infarction, termed pituitary apoplexy (another emergent condition), may result in not only diplopia, but also sudden vision loss and headache.26
Cases of painful ophthalmoplegia that exhibit enhancement of the cavernous sinus on neuroimaging and respond to steroids are often misdiagnosed as Tolosa-Hunt syndrome. A diagnosis of idiopathic Tolosa-Hunt syndrome should be considered only after ruling out neoplasm, infection and other inflammatory etiologies.27
Cavernous sinus aneurysm—Within the cavernous sinus, the internal carotid artery (ICA) is surrounded by the sympathetic fibers. Additionally, unlike CN III and CN IV, which lie in the lateral wall of the cavernous sinus (along with V1 and V2), CN VI is proximal to the carotid artery. Thus, an abduction deficit with ipsilateral Horner’s syndrome may lead you to suspect an intracavernous ICA aneurysm. Aneurysmal rupture is uncommon and typically not life threatening (unlike aneurysm in the subarachnoid space), but may cause a carotid cavernous fistula.
Carotid cavernous fistula—Direct carotid cavernous fistulas occur with communication between the arterial and venous system within the cavernous sinus, often from trauma or aneurysm rupture. The venous system is unable to drain the high-pressure influx of blood from the arterial system, leading to acute cavernous sinus syndrome, and possibly proptosis, chemosis and pulsatile exophthalmos. Indirect fistulas have a more insidious onset and tend to occur spontaneously.
As demonstrated, CN palsy localizing to the subarachnoid space or cavernous sinus may indicate a medical emergency, requiring immediate hospitalization. Newer steady-state free precession (SSFP) MRI Images sequences (e.g., FIESTA/CISS) are best at imaging the CNs in the subarachnoid space.28
A common cause of isolated CN palsies occurring in patients older than 50 years is microvascular ischemia (i.e., diabetes and hypertension), which should resolve within three to six months. Recent research recommends all patients presenting with acute, isolated ocular motor neuropathies undergo additional work-up.29,30
In addition, any diplopia or ocular misalignment in a patient older than 50 years should prompt concern for giant cell arteritis (GCA), and urgent ESR, c-reactive protein and CBC is recommended.24
Lab Testing for Thyroid Eye Disease• TSH• T3 • T4 • Thyroperoxidase antibodies • Thyroglobulin antibodies • Thyroid stimulating immunoglobulin |
Evaluating for EOM Disorders Stemming from the Neuromuscular Junction
If you do not find any features that definitively localize to the brain or a specific point along a CN, the next step is to consider a disease process of the neuromuscular junction (NMJ). Myasthenia gravis (MG), which often mimics CN palsies, is the most common NMJ disorder.19
In cases of MG, waves of depolarization arrive at the presynaptic neuron terminal initiating the release of acetylcholine, similar to that in patients without MG. However, in MG, as acetylcholine makes its way across the synaptic cleft it is unable to fuse with its receptor due to the presence of antibodies. These antibodies may affect the receptor in three ways; antibodies may:1. Bind to the receptor and initiate an inflammatory reaction.
2. Block the receptor and simply prevent acetylcholine attachment.
3. Modulate by cross-linking, thereby causing engulfment of the receptor.
Therefore, acetylcholine is unable to initiate contraction of the intended muscle cell. In a smaller percentage of patients, antibodies are produced against the Muscle Specific Kinase (MuSK) receptor, which is responsible for regulating the population of acetylcholine receptors.31 In both cases, the receptor is the limiting factor. However, if the availability of the acetylcholine is also decreased, the effects are compounded. Consequently, MG is characterized by patients who have weakness, which worsens with fatigue.
Myasthenia Gravis Testing Techniques
|
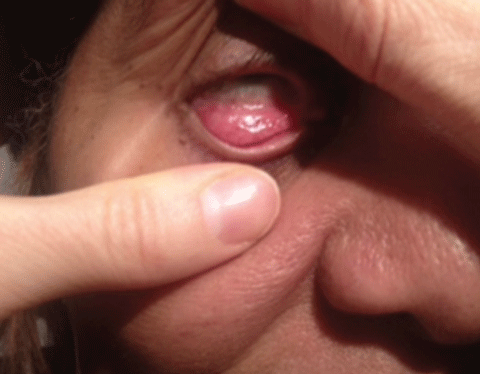
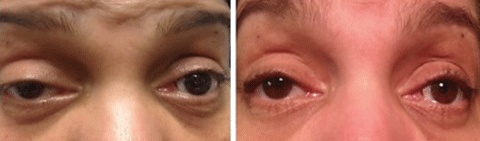
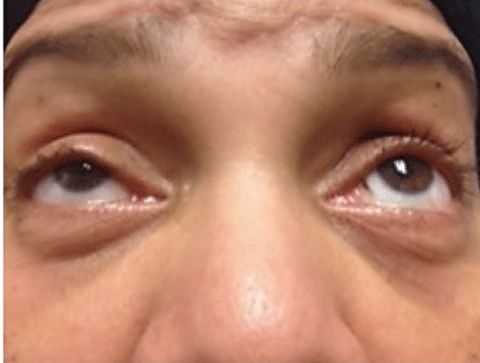
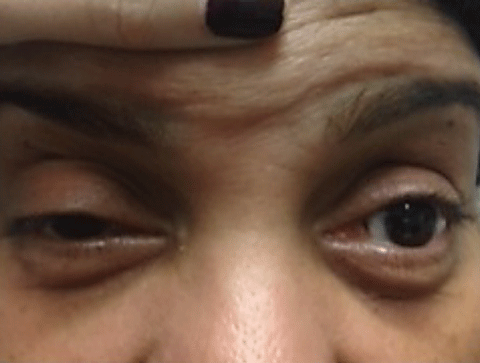
The majority of patients with MG present with ptosis and ocular misalignment. To explain the increased susceptibility of the muscles within and around the orbit, investigators propose that the acetylcholine receptors are antigenically different in this area.32 MG is subdivided into ocular MG, which is isolated to the ocular muscles and generalized MG, which involves head, limb and respiratory muscles. Patients with ocular MG have roughly a 50% chance of developing generalized MG within two years of onset.33
Diagnosis of MG—In all patients with diplopia or ocular misalignment, regardless of the pattern, myasthenia gravis must be in your differential diagnosis. Three in-office tests aid in the diagnosis of MG (above). These tests check for an increased ptosis after fatigue, improvement in ptosis when acetylcholinesterase enzyme is deactivated by cold, and general weakness of the orbicularis muscles.
To further confirm the suspected diagnosis, test serologically for the acetylcholine receptor antibodies (binding, blocking and modulating), and for the less common Anti-MusK antibodies. If these fail to confirm a diagnosis, a more sensitive test for MG is the single fiber electromyogram (sfEMG), which employs an electrode to record the action potentials of individual muscle cells. If testing for ocular MG, the sfEMG should be performed on the frontalis or orbicularis oculi muscle. A positive result shows increased jitter.
Patients diagnosed with systemic or ocular MG need a chest CT to rule out thymoma. Referral can then be made to neurology/neuro-ophthalmology for systemic treatment that may include cholinesterase inhibitors, immunosuppressants and corticosteroids. A careful review of medications is necessary as there are many which can incite or worsen MG, these include statins, antibiotics, anticonvulsants, antiarrhythmics, beta blockers (both systemic and topical) and botulinum toxin.
EOM Disorders Stemming From the Orbit
Clinically, two tests can localize diplopia to the orbit. They are exophthalmometry and forced duction testing. The eye can either be exophthalmic or enophthalmic, depending on the cause. A positive forced duction test (when the eye is unable to be manually moved in the direction of limited gaze) suggests orbital localization. There may be associated eye pain, injection or chemosis, as well as features to suggest afferent visual system compromise. If an orbital cause for diplopia is suspected, referral to an orbital/oculoplastic specialist is often warranted.
Orbital fracture—Blowout fractures of the orbit are commonly caused by blunt trauma and frequently cause diplopia. EOM restriction in the setting of recent trauma should cause suspicion for orbital fracture. Most commonly, the orbital floor is affected, causing entrapment of the inferior rectus and subsequent supraduction deficit. Since the infraorbital nerve runs through the inferior orbital groove, hypoesthesia of the lower eyelid increases suspicion of an orbital floor fracture. Enophthalmos is seen in floor fractures if orbital contents sublux into the maxillary sinus. Less commonly, the medial rectus can become entrapped with medial wall fractures, as the ethmoid is the thinnest bone of the orbit. An orbital CT allows visualization of the fracture.34
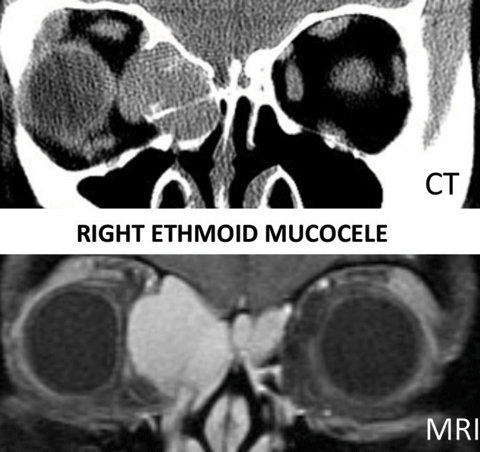 |
| Diplopia combined with proptosis, pain, chronic sinusitis or history of endoscopic surgery should prompt suspicion of mucocele. |
Mucocele—Mucus lines the paranasal sinuses adjacent to the orbits. When there is scarring and obstruction of the sinus ostium, a mucocele can develop, eroding the bony sinus wall and invading the brain and orbit with potential for abscess and rupture. Diplopia combined with proptosis, pain, chronic sinusitis or history of endoscopic surgery should prompt suspicion of mucocele. Work-up includes neuroimaging of the orbits; CT will assess for bony destruction.34,35
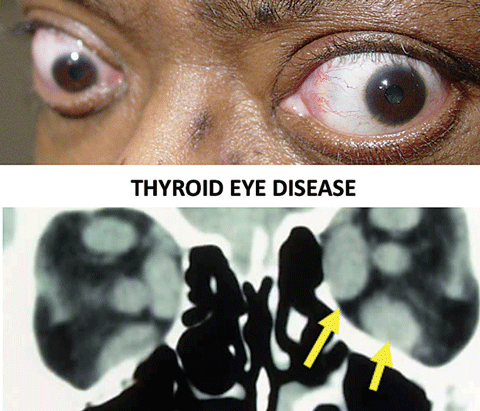 |
| Thyroid eye disease is the most common cause of ocular motility restrictions in adults. The arrows show enlarged bellies of the extraocular muscles. Click image to enlarge. |
Thyroid eye disease—Thyroid eye disease is the most common cause of motility restriction in adults, involving inflammation of the EOMs.36,37 The inferior rectus (supraduction deficit) and medial rectus (abduction deficit) are most commonly affected. Exophthalmos, eyelid retraction and eyelid edema are also possible, but not necessarily present. Positive forced duction testing and increased intraocular pressure in affected gazes supports the diagnosis. Work-up for thyroid eye disease includes lab testing (thyroid functions and antibodies) and neuroimaging of the orbit (CT or MRI) looking for extraocular muscle belly enlargement.36-41
Other Conditions with Orbital Mass Effect
Inflammatory Conditions—The spectrum of orbital inflammatory disease ranges from nonspecific inflammation of one orbital structure to widespread inflammation from systemic disease. Usually, patients present with diplopia (any pattern possible), proptosis, eye pain and possibly eyelid edema, chemosis or injection. A thorough history regarding onset, associated symptoms and systemic health conditions is critical in guiding differentials.
Idiopathic orbital inflammatory syndrome—IOIS can affect many structures of the orbit and is a diagnosis of exclusion. It commonly presents with unilateral symptoms with variable degree of inflammation, fibrosis and mass effect based on the structure involved. This condition should improve quickly with corticosteroids. If not, or in cases of recurrence, alternate etiologies should be considered.39-41
Sarcoidosis—This condition can affect any part of the orbit. Although it usually presents with enlargement of the lacrimal gland(s), it can also manifest as a diffuse, solid mass with infiltration of orbital fat, lacrimal sac or extraocular muscles. Abnormal skin lesions, chronic cough, shortness of breath or uveitis may raise suspicion for sarcoidosis. Although a mass may be seen on neuroimaging, or angiotensin converting enzyme may be positive, definitive diagnosis is made by biopsy, often of the lacrimal gland or lung.42,43
Granulomatosis with polyangiitis (GPA)(formerly Wegener’s)—Ocular manifestations can occur with or without symptoms of systemic GPA, although they are more common later in the disease course. Ocular findings are commonly associated with adjacent paranasal sinus or nasal granulomatous disease. History of chronic sinusitis, rhinitis or epistaxis or a saddle nose deformity indicate possible GPA.44,45
Lab Testing for Orbital InflammationsSarcoid• ACE GPA • C-ANCA • P-ANCA GCA • ESR • C-reactive protein • CBC |
Giant cell arteritis (GCA)—Orbital inflammatory disease is an uncommon yet underdiagnosed finding in GCA despite being the most common vasculitis in patients older than 50. In addition to symptoms of orbital inflammatory disease, these patients might present with other classic features of GCA: headache, malaise, weight loss and vision loss.46-47
Infectious conditions—Orbital infections include orbital cellulitis, subperiosteal or orbital abscess. Bacterial infections are more common than fungal, but both etiologies are possible. In addition to the features found in inflammatory orbital disorders, infectious causes may present with fever or secondary infection. The most common bacterial causes of orbital infections are Staphyococcus aureus, Streptococcus pneumoniae and anaerobic Gram negative bacilli, such as Prevotella, Porphyromonas, and Fusobacterium. In addition, mucormycosis, aspergillosis and Mycobacterium tuberculosis have been implicated, more commonly in immunocompromised hosts.34,48-49
Cancers
Primary and secondary orbital malignancy must be considered in the setting of persistent progression or inflammation despite treatment.
Lymphoma—Lymphocytic lesions of the orbit include both benign lymphoid hyperplasia and malignant lymphoma, which is almost exclusively B-cell lymphoma. These usually manifest in the orbit as painless masses; they may affect specific structures such as the lacrimal gland. Painful lymphomas are rare, tend to progress more rapidly and have worse prognosis. MRI demonstrates a well-defined soft-tissue mass affecting, but not destroying, surrounding tissues. Biopsy is required to confirm the diagnosis.34,50
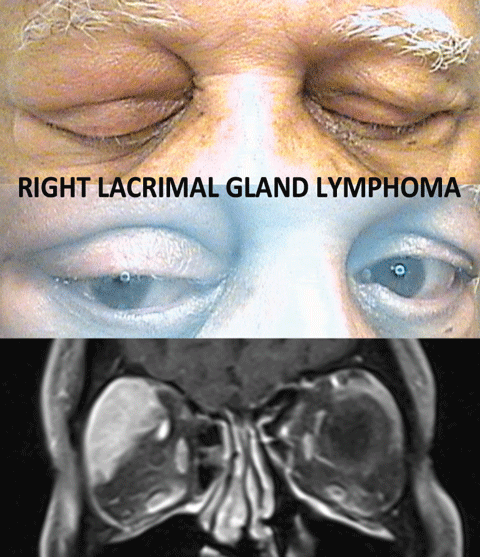 |
| Lymphomatic lesions usually manifest in the orbit as painless masses that affect specific structures, such as the lacrimal gland, as noted in this patient. The MRI demonstrates a well-defined soft-tissue mass affecting, but not destroying, surrounding tissue. Click image to enlarge. |
Metastases—These can occur to specific orbital structures, including the EOMs. When an isolated muscle is enlarged in the setting of a history of cancer, consider metastasis before jumping to a diagnosis of thyroid eye disease. Proptosis is common when mass effect is present. However, there is also possibility of enophthalmos if the metastasis causes fibroblast contraction and globe retraction. This can be seen with scirrhous breast cancer, among others. Consider mammogram, colonscopy and imaging of the chest, abdomen and pelvis in any unexplained non-traumatic enophthalmos.34,50-51
A step-wise approach will increase your comfort level in dealing with diplopia and ocular motility abnormalities. Use your history and clinical examination to localize the lesion to the brain, nerve, junction or orbit. This will streamline the process and help you understand potential urgency, necessary work-up and the most appropriate referral.
Dr. Malloy is the director of the Neuro-ophthalmic Disease Service at Pennsylvania College of Optometry, Salus University.
Dr. Draper teaches neuro-ophthalmic disease at Salus University and practices in the Neuro-Ophthalmic Disease and Low Vision Specialty Services at The Eye Institute.
Drs. Maglione and Moody are in the process of completing the new two-year advanced residency program in neuro-ophthalmic disease at Salus under the direction of
Drs. Malloy and Draper.
|
1. Malloy K. Neuro-ophthalmic disease basics: Focus on the efferent visual system evaluation. Rev Optom. 2015. Feb;152:74-83. 2. Bourne S, Duhaime A. Transient horizontal gaze palsy in a one-month-old boy after a fall. Pediatr Neurosurg. 2016;51(1):42-7. 3. Eggenberger E. Supranuclear eye movement abnormalities. Continuum (Minneap Minn). 2014 Aug;20(4 Neuro-ophthalmology):981-92. 4. Strupp M, Kremmyda O, Adamczyk C, et al. Central ocular motor disorders, including gaze palsy and nystagmus. J Neurol. 2014 Sep;261 Suppl 2:S542-58. 5. Koskas P, Heran F. Towards understanding ocular motility: III, IV, and VI. Diagnostic and interventional imaging. 2013; 94: 1017-1031. 6. Brazis P. Isolated palsies of cranial nerves III, IV, and VI. Semin Neurol. 2009;29:14-28. 7. Bronstein A, Rudge P, Gresty M, et al. Abnormalities of horizontal gaze. Clinical oculographic and magnetic resonance imaging findings. II gaze palsy and internuclear ophthalmoplegia. Journal of Neurology, Neurosurgery and Psychiatry. 1990;53:20-207. 8. Gutowski N, Chilton J. The congenital cranial dysinnervation disorders. Arch Dis Child. 2015 Jul;100(7):678-81. 9. Xia S, Li R, Li Y, et al. MRI findings in Duane’s ocular retraction syndrome. Clin Radiol. 2014 May;69(5):e191-8. 10. Oystreck D. Congenital and genetic ocular motility disorders: update and considerations. Am Orthopt J. 2015;65:58-66. 11. Gnanapavan S, Sillery E, Acheson J, Toosy A. Parinaud’s syndrome—A rare presentation of clinically isolated syndrome. Mult Scler Relat Disord. 2014 May;3(3):398-401. 12. Alemdar M, Kamaci S, Budak F. Unilateral midbrain infarction causing upward and downward gaze palsy. J Neuroophthalmol. 2006 Sep;26(3):173-6. 13. Eggenberger E. Eight and-a-half syndrome: one-and-a-half syndrome plus cranial nerve VII palsy. J Neuroophthalmol 1998;18(2) 114-116. 14. Rosini F, Pretegiani E, Guideri F, Cerase A, Rufa A. Eight and a half syndrome with hemiparesis and hemihypesthesia: the nine syndrome? J Stroke Cerebrovasc Dis. 2013 Nov;22(8):e637-8. 15. Connors R, Ngan V, Howard J. A case of complete lateral gaze paralysis and facial diplegia: the 16 syndrome. J Neuroophthalmol. 2013 Mar;33(1):69-70. 16. Lee H, de Kort PL. 16 syndrome in a patient with multiple sclerosis. J Neuroophthalmol. 2013 Jun;33(2):203-4. 17. Howard V, Barrow D. Neuro-ophthalmology and intracranial aneurysms. World neurosurg. 2015 Mar;83(3):291-3. 18. Ramgopal S, Obeid R, Zuccoli G, et al. Lyme-disease-related intracranial hypertension in children: clinical and imaging findings. J Neurol. 2016 Mar;263(3):500-7. 19. Neil R. Miller NR, Newman NJ, Biousse V, Kerrison JB, Editors. Walsh and Hoyts Clinical Neuro-ophthalmology. Lippincott Williams & Wilkins, Philadelphia, 2005. 20. Umansky F, Elidan J, Valarezo A. Dorello’s Canal: a microanatomical study. J Neurosurg. 1991 Aug;75(2):294-8. 21. Malloy KA. Prostate cancer metastasis to clivus causing cranial nerve VI palsy. Optometry. 2007 Feb;78(2):55-62. 22. Adams ME, Linn J, Yoursry, I. Pathology of the ocular motor nerve III, IV and VI. Neuroimag Clin N Am. 2008 May;18(2):261-82. 23. Biousse V, Newman N. Aneurysms and subarachnoid hemorrhage. Neurosurg Clin N Am. 1999 Oct;10(4):631-51. 24. Biousse V and Newman N. Neuro-ophthalmology Illustrated. Thieme Medical Publishers, Inc. New York. 614pp. 25. Sindou M, Nebbal M, Guclu B. Cavernous sinus meningiomas: imaging and surgical strategy. Adv Tech Stand Neurosurg. 2015: 42:103-21. 26. Simon S., Torpy D, Brophy B, Blumbergs P, Selva D, Crompton JL. Neuro-ophthalmic manifestations and outcomes of pituitary apoplexy—alife and sight-threatening emergency. N Z Med J. 2011 May 27;124(1335):52-9. 27.İlgen Uslu F, Özkan M. Painful ophthalmoplegia: a case report and literature review. Agri. 2015;27(4):219-23. 28. Sheth S, Branstetter BF 4th, Escott EJ. Appearance of normal cranial nerves on steady-state free precession MR images. Radiographics. 2009 Jul-Aug;29(4):1045-55. 29. Kung NH, Van Stavern GP. Isolated ocular motor nerve palsies. Semin Neurol. 2015 Oct;35(5):539-48. 30. Tamhankar MA, Volpe NJ. Management of acute cranial nerve 3, 4 and 6 palsies: role of neuroimaging. Curr Opin Ophthalmol. 2015 Nov;26(6):464-8. 31. Hoch, W. McConville J, Helms S, et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nature Medicine. 2001;7:365-8. 32. Kaminski H, Zhuyi L, Richmonds C, et al. Susceptibility of ocular tissues to autoimmune diseases. Ann N Y Acad Sci. 2003;998:362-74. 33. Patil-Chhablani, P, Devendrav V, Rashminanilkumar G, Akshaygopinathan N. Ocular myasthenia gravis: A review. Indian J Ophthalmol 2014; 62(1): 985. 34. Rose G, Verity D. Neuro-ophthalmology of orbital disease. Handbook of Clinical Neurology. 2011;102(3):467-91. 35. Venugopal M, Sagesh M. Proptosis: The ENT surgeon’s perspective. Indian J Otolaryngol Head Neck Surg. 2013. Aug;65(Suppl 2):247-50. 36. Barrio-Barrio J, Sabater A, Bonet-Farriol E, et al. Graves’ Ophthalmopathy: VISA versus EUGOGO classification, assessement, and management. Journal of Ophthalmology. 2015 Feb;2015:1-16. 37. McAlinden C. An overview of thyroid eye disease. Eye Vis (Lond). 2014;1(9):1-4. 38. Lutt J, Lim L, Phal P, et al. Orbital inflammatory disease. Semin Arthritis Rheum. 2008; 37:207-222. 39. Costa R, Dumitrascu O, Gordon L. Orbital myositis: diagnosis and management. Current Allergy and Asthma Reports. 2009;9:316-23. 40. Pakdaman M, Sepahdari A, Elkhamary S. Orbital inflammatory disease: pictorial review and differential diagnosis. World J Radiol. 2014; 6(4):106-15. 41. Gordon L. Orbital inflammatory disease: a diagnostic and therapeutic challenge. Eye. 2006;20:1196-206. 42. Demirci H, Christianson M. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol. 2011;151:1074-80. 43. Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Opthalmol 2007;144:769-75. 44. Holle J, Voigt C, Both M, et al. Orbital masses in granulomatosis with polyangiitis are associated with refractory course and a high burden of local damage. Rheumatology. 2013;52:875-82. 45. Provenzale J, Mukherji S, Allen N, et al. Orbital involvement by Wegener’s granulomatosis: imaging findings. AJR. 1996;166:929-34. 46. Lee A, Tang R, Feldon S, et al. Orbital presentations of giant cell arteritis. Graefe’s Arch Clin Exp Ophthalmol. 2001;239:509-13. 47. Gonzalez-Gay M, Carros S, Lopez-Diaz M, et al. Giant cell arteritis. Medicine. 2005;84:269-76. 48. Warburton R, Brookes C, Golden B, et al. Orbital apex disorders: a case series. Int J Oral Maxillofac Surg. 2015;Oct:1-10. 49. Kirszrot J, Rubin PAD. Invasive fungal infections of the orbit. International Ophthalmology Clinics. 2007; 47(2):117-132. 50. Bonavolonta G, Strianese D, Grassi, P, et al. An analysis of 2,480 space-occupying lesions of the orbit from 1976 to 2011. Ophthal Plast Reconstr Surg. 2013; 29:79-86. 51. Ahmad S, Esmaeli B. Metastatic tumors of the orbit and ocular adnexa. Curr Opin Ophthalmol. 2007;18:405-13. 52. Manley D, Alvi R. Brown’s syndrome. Curr Opin Ophthalmol. 2011 Sep;22(5):432-40. 53. Currie S, Goddard T. MR imaging features of acquired Brown syndrome. AJNR. 2009 Oct;30:1778-9. |